причины возникновения, подходы к диагностике и лечению » Акушерство и Гинекология
ФГБУ Эндокринологический научный центр Минздравсоцразвития России, Москва, Россия; ГБУЗ МО Московский Областной научно-исследовательский клинический институт им. М.Ф. Владимирского, Москва, Россия
Формирующийся в результате нарушения синтеза и/или секреции гонадотропинов гипогонадотропный гипогонадизм может быть органическим (т.е. формирующимся в результате анатомо-функциональных расстройств гипоталамо-гипофизарной области при различных новообразованиях хиазмально-селлярной локализации) или функциональным, приобретенным или врожденным (мутации гена рецептора ГнРГ, β-субъединицы ФСГ; генов SF-1, DAX-1, нарушения в генах рецепторов ЛГ и ФСГ и пр.). В формировании функциональных нарушений принимают участие нейропептид Y (NPY), кортикотропин-рилизинг-гормон (КРГ), лептин, грелин и β-эндорфин. Диагностика заболевания включает определение гонадотропинов, исключение гиперпролактинемии и других эндокринных расстройств, ультразвуковое исследование матки и яичников, магнитно-резонансную томографию головного мозга. При длительности аменореи более года показано исследование плотности костной ткани. При выявлении причин гипогонадотропного гипогонадизма в первую очередь необходимо их устранение (например, ликвидация стресса, исключение физических нагрузок, восстановление нормальной массы тела, или лечение опухолей гипофиза и т.д.), что может приводить к восстановлению менструальной функции и фертильности. Однако в большинстве случаев при стойкой гипоэстрогении у пациенток с гипогонадотропным гипогонадизмом показана эстроген-гестагенная терапия в циклическом режиме, для длительного применения эффективной и безопасной является комбинация 17β-эстрадиола 2 мг и дидрогестерона 10 мг.
гипогонадизм
гипоэстрогенемия
эстроген-гастагенная терапия
1. Иловайская И.А. Этиология приобретенного гипопитуитаризма у взрослых. Доктор Ру. 2010: 7 (58, ч. 2): 8–13.
2. De Bellis A., Ruocco G., Battaglia M., Conte M., Coronella C., Tirelli G. et al. Immunological and clinical aspects of lymphocytic hypophysitis. Clin. Sci. (Lond). 2008; 114(6): 413–21.
3. Bondanelli M., De Marinis L., Ambrosio M.R., Monesi M., Valle D., Zatelli M.C. et al. Occurrence of pituitary dysfunction following traumatic brain injury. J. Neurotrauma. 2004; 21(6): 685–96.
4. Meczekalski B., Podfigurna-Stopa A., Warenik-Szymankiewicz A., Genazzani A.R. Functional hypothalamic amenorrhea: current view on neuroendocrine aberrations. Gynecol. Endocrinol. 2008; 24(1): 4–11.
5. Marshall L. A. Clinical evaluation of amenorrhea in active and athletic women. Clin. Sports Med. 1994; 13: 371–87.
6. Berga S.L., Loucks-Daniels T.L., Adler L.J., Chrousos G.P., Cameron J.L., Matthews K.A., Marcus M.D. Cerebrospinal fluid levels of corticotropin-releasing hormone in women with functional hypothalamic amenorrhea. Am. J. Obstet. Gynecol. 2000; 182(4): 776–81; discussion 781–4.
7. Genazzani A.D. Neuroendocrine aspects of amenorrhea related to stress. Pediatr. Endocrinol. Rev. 2005; 2(4): 661–8.
8. Rivier C., Brownstein M., Spiess J., Rivier J., Vale W. In vivo corticotropin-releasing factor-induced secretion of adrenocorticotropin, beta-endorphin, and corticosterone. Endocrinology. 1982; 110(1): 272–78.
9. Moschos S., Chan J.L., Mantzoros C.S. Leptin and reproduction: a review. Fertil. Steril. 2002; 77: 433–44.
10. Ahima R.S. Body fat, leptin, and hypothalamic amenorrhea. N. Engl. J. Med. 2004; 351: 959–62.
11. Carro E., Pinilla L., Seoane L.M., Considine R.V., Aguilar E., Casanueva F.F., Dieguez C. Influence of endogenous leptin tone on the estrous cycle and luteinizing hormone pulsatility in female rats. Neuroendocrinology. 1997; 66(6): 375–7.
12. Herpertz S., Albers N., Wagner R., Pelz B., Köpp W., Mann K. et al. Longitudinal changes of circadian leptin, insulin and cortisol plasma levels and their correlation during refeeding in patients with anorexia nervosa. Eur. J. Endocrinol. 2000; 142(4): 373–9.
Eur. J. Endocrinol. 2000; 142(4): 373–9.
13. Andrico S., Gambera A., Specchia C., Pellegrini C., Falsetti L., Sartori E. Leptin in functional hypothalamic amenorrhoea. Hum. Reprod. 2002; 17(8): 2043–8.
14. Welt C.K., Chan J.L., Bullen J., Murphy R., Smith P., DePaoli A.M. et al. Recombinant human leptin in women with hypothalamic amenorrhea. N. Engl. J. Med. 2004; 351(10): 987–97.
15. Pedrazzini T., Pralong F., Grouzmann E. Neuropeptide Y: the universal soldier. Cell. Mol. Life Sci. 2003; 60: 350–77.
16. Wójcik-Gładysz A., Polkowska J. Neuropeptide Y — a neuromodulatory link between nutrition and reproduction at the central nervous system level. Reprod. Biol. 2006; 6 (Suppl. 2): 21–8.
17. Cummings D.E., Weigle D.S., Frayo R.S., Breen P.A., Ma M.K., Dellinger E.P., Purnell J.Q. Plasma ghrelin levels after diet-induced weight loss or gastric bypass surgery. N. Engl. J. Med. 2002; 346(21): 1623–30.
18. Schneider L.F., Warren M.P. Functional hypothalamic amenorrhea is associated with elevated ghrelin and disordered eating. Fertil. Steril. 2006; 86(6): 1744–9.
19. Tolle V., Kadem M., Bluet-Pajot M.T., Frere D., Foulon C., Bossu C. et al. Balance in ghrelin and leptin plasma levels in anorexia nervosa patients and constitutionally thin women. J. Clin. Endocrinol. Metab. 2003; 88(1): 109–16.
20. Schneider L.F., Warren M.P. Functional hypothalamic amenorrhea is associated with elevated ghrelin and disordered eating. Fertil. Steril. 2006; 86(6): 1744–9.
21. Chan J.L., Bullen J., Lee J.H., Yiannakouris N., Mantzoros C.S. Ghrelin levels are not regulated by recombinant leptin administration and/or three days of fasting in healthy subjects. J. Clin. Endocrinol. Metab. 2004; 89(1): 335–43.
22. De Souza M.J., Leidy H.J., O’Donnell E., Lasley B., Williams N.I. Fasting ghrelin levels in physically active women: relationship with menstrual disturbances and metabolic hormones. J. Clin. Endocrinol. Metab. 2004; 89(7): 3536–42.
23. Marcus M. D., Loucks T.L., Berga S.L. Psychological correlates of functional hypothalamic amenorrhea. Fertil. Steril. 2001; 76(2): 310–6.
D., Loucks T.L., Berga S.L. Psychological correlates of functional hypothalamic amenorrhea. Fertil. Steril. 2001; 76(2): 310–6.
24. Bry-Gauillard H., Trabado S., Bouligand J., Sarfati J., Francou B., Salenave S. et al. Congenital hypogonadotropic hypogonadism in females: clinical spectrum, evaluation and genetics. Ann. Endocrinol. (Paris). 2010; 71(3): 158–62.
25. Tsutsumi R., Webster N.J. GnRH pulsatility, the pituitary response and reproductive dysfunction. Endocr. J. 2009; 56(6): 729–37.
26. Зыряева Н.А. Системные изменения и их коррекция при гипогонадотропном гипогонадизме у женщин: Автореф. дис. … канд. мед. наук. М.; 2004. 29 с.
27. Иловайская И.А., Зекцер В.Ю., Пищулин А.А., Гончаров Н.П., Марова Е.И. Эффективность заместительной гормональной терапии в лечении женщин с гипогонадотропным гипогонадизмом. Акушерство и гинекология. 2005; 4: 53–4.
28. Зекцер В.Ю., Иловайская И.А., Ильин А.В., Колесникова Г.С., Марова Е.И., Мельниченко Г.А. Нарушения метаболизма и их коррекция у женщин с гипогонадотропной аменореей. Ожирение и метаболизм. 2006; 4: 54–7.
29. Иловайская И.А. Диагностика и лечение гипопитуитаризма. В кн.: Дедов И.И., ред. Клиническая нейроэндокринология. М.: УП Принт; 2011: 220–38.
30. Дедов И.И., Марова Е.И., Иловайская И.А., Манченко О.В. Гипогонадотропный гипогонадизм у женщин. Акушерство и гинекология. 2001; 3: 6-10.
31. Иловайская И.А., Зекцер В.Ю., Ильин А.В., Гончаров Н.П., Дедов И.И. Эффекты длительного применения эстроген-гестагенной терапии у женщин репродуктивного возраста с изолированным гипогонадотропным гипогонадизмом. Ожирение и метаболизм. 2010; 1: 52–7.
32.Дедов И.И., Мельниченко Г.А., Романцова Т.И., Дзеранова Л.К. и др. Гиперпролактинемия. Современные подходы и старые проблемы. Вестник репродуктивного здоровья. 2009; 2: 2–8.
33.Иловайская И.А., Зекцер В.Ю., Михайлова Д.С., Гончаров Н.П., Мельниченко Г.А. Влияние заместительной гормональной терапии на качество жизни пациенток с гипогонадотропным гипогонадизмом. Вопросы гинекологии, акушерства и перинатологии. 2008; 7(3): 48–55.
Вопросы гинекологии, акушерства и перинатологии. 2008; 7(3): 48–55.
34.Seeger H., Mueck A.O. Effects of dydrogesterone on the vascular system. Gynecol. Endocrinol. 2007; 23 (Suppl. 1): 2–8.
Дедов Иван Иванович, доктор медицинских наук, профессор, академик РАН и РАМН, директор ФГБУ Эндокринологический научный центр Минздравсоцразвития РФ, директор Федерального диабетологического центра Минздравсоцразвития РФ, заведующий кафедрой эндокринологии Первого МГМУ им. И.М. Сеченова, президент РАМН, лауреат премии и золотой медали им. Н.И. Пирогова, заслуженный деятель науки РФ
Адрес: 117036, Россия, Москва, ул. Дм. Ульянова, д. 11. Телефон: 8 (499) 124-43-00. E-mail: [email protected]
Мельниченко Галина Афанасьевна, доктор медицинских наук, профессор, заместитель директора по научной работе ФГБУ Эндокринологический научный центр Минздравсоцразвития России, директор Института клинической эндокринологии Эндокринологического научного центра, академик РАМН
Адрес: 117036, Россия, Москва, ул. Дм. Ульянова, д. 11. Телефон: 8 (495) 500-00-96. E-mail: [email protected]
Иловайская Ирэна Адольфовна, кандидат медицинских наук, доцент, ведущий научный сотрудник отделения терапевтической эндокринологии ГБУЗ МО Московский областной научно-исследовательский клинический институт (МОНИКИ) им. М.Ф. Владимирского
Адрес: 129110, Россия, Москва, ул. Щепкина, д. 61/2, корпус 9. Телефон: 8 (901) 545-38-09. E-mail: [email protected] [email protected]
Лечение мужского гипогонадизма препаратами тестостерона | Манушарова Р.А., Черкезова Э.И.
Мужской гипогонадизм обусловлен недостаточной секрецией тестостерона или отсутствием эндогенного тестостерона, который может возникнуть при многих заболеваниях, связанных с патологией яичников (первичная тестикулярная недостаточность) или патологией в регулирующих центрах (гипогонадотропный гипогонадизм). Вторичный, или гипогонадотропный гипогонадизм возникает вследствие гонадотропной недостаточности, которая может сочетаться с недостаточностью других тропных гормонов гипофиза. Секреция тестостерона контролируется гипоталамо–гипофизарно–яичковой системой [1]. Выделение из гипоталамуса рилизинг–гормона ЛГ (ЛГРГ) стимулирует выброс гонадотропинов (ЛГ, ФСГ) из гипофиза. В мужском организме ЛГ стимулирует синтез тестостерона клетками Лейдига и способствует развитию яичек, а ФСГ совместно с тестостероном регулирует сперматогенез и созревание сперматозоидов [2]. ФСГ также повышает активность ЛГ и синтез тестостерона [2,3]. В свою очередь, тестостерон регулирует выделение ЛГ и ФСГ из гипофиза посредством отрицательной обратной связи. В клетках Сертоли, находящихся в яичках, вырабатывается полипептид – ингибин, который тормозит секрецию ФСГ (продукция последней стимулируется активином).
Секреция тестостерона контролируется гипоталамо–гипофизарно–яичковой системой [1]. Выделение из гипоталамуса рилизинг–гормона ЛГ (ЛГРГ) стимулирует выброс гонадотропинов (ЛГ, ФСГ) из гипофиза. В мужском организме ЛГ стимулирует синтез тестостерона клетками Лейдига и способствует развитию яичек, а ФСГ совместно с тестостероном регулирует сперматогенез и созревание сперматозоидов [2]. ФСГ также повышает активность ЛГ и синтез тестостерона [2,3]. В свою очередь, тестостерон регулирует выделение ЛГ и ФСГ из гипофиза посредством отрицательной обратной связи. В клетках Сертоли, находящихся в яичках, вырабатывается полипептид – ингибин, который тормозит секрецию ФСГ (продукция последней стимулируется активином).
Первичный гипогонадизм обусловлен генетическими причинами или аномалией развития (синдром Клайнфельтера; 47 хху и его варианты), или вследствие приобретенных заболеваний (вирусный орхит и др.).
Вторичный, или гипогонадотропный гипогонадизм имеет наследственную (синдром Каллманна) или приобретенную (опухоли гипофиза) природу. Большинство форм гипогонадизма, диагностируемых у взрослых, являются приобретенными и наиболее часто развиваются вследствие ожирения, тяжелых системных заболеваний, приема некоторых лекарств [4]. Часто вторичный гипогонадизм развивается как результат острых и хронических заболеваний, при синдроме приобретенного иммунодефицита, серповидно–клеточной анемии, циррозе печени, почечной недостаточности. Причиной вторичного гипогонадизма могут быть эпидемиологические факторы (курение, прием алкоголя), возрастные изменения в гипоталамусе и гипофизе.
У мужчин возрастные изменения в репродуктивной системе развиваются медленнее, чем у женщин. В настоящее время установлено, что первые изменения возникают на уровне яичек и заключаются в снижении числа клеток Лейдига и метаболической активности ферментов, участвующих в синтезе тестостерона, что приводит к уменьшению продукции тестостерона в ответ на стимуляцию гонадотропинaми и снижению его биодоступности.
Клинические проявления гипогонадизма слабо выражены, и поэтому мужской гипогонадизм считается плохо диагностируемым заболеванием.
Клиническая манифестация гипогонадизма зависит от возраста, в котором он возник, а также его причины и длительности недостаточности тестостерона. При развитии гипогонадизма во взрослом состоянии чаще всего не удается выявить изменений, но в некоторых случаях обнаруживается умеренная гинекомастия, уменьшение количества волос на лице и теле, при этом яички пальпаторно мягкие и небольших размеров [4,5].
Клинические проявления дефицита тестостерона у взрослых мужчин характеризуются рядом признаков и симптомов:
• Снижением полового влечения
• Ухудшением настроения
• Эректильной дисфункцией
• Олигоспермией или азооспермией
• Снижением плотности костной ткани
• Регрессией вторичных половых признаков
• Потерей мышечной массы и мышечной силы
• Усталостью
Возрастная недостаточность тестостерона может сочетаться с некоторыми физиологическими изменениями, такими как снижение мышечной массы и силы, увеличение количества жировой ткани, снижение костной массы и повышение частоты остеопороза и переломов даже при незначительной травме, снижение либидо и повышение частоты нарушений эрекции, а также ухудшение общего самочувствия.
При постановке диагноза необходимо определение в крови уровня тестостерона и гонадотропинов. При гипогонадизме отмечается постоянно низкая концентрация тестостерона в сочетании с повышенным (первичный гипогонадизм) или сниженным (вторичный гипогонадизм) уровнем ЛГ и ФСГ в крови.
Тестостерон – преобладающий гормон в крови мужчины, вырабатывается преимущественно (95%) яичками, образуется из холестерина, в значительно меньших количествах – корой надпочечников. Ежесуточный объем выделения тестостерона в плазму крови составляет приблизительно 6 мг, в яичках откладывается лишь незначительное количество тестостерона. Поступая в кровоток, тестостерон транспортируется в плазме при помощи секс–стероидсвязывающего глобулина (СССГ) или в связанном с альбумином или с другими белками крови виде.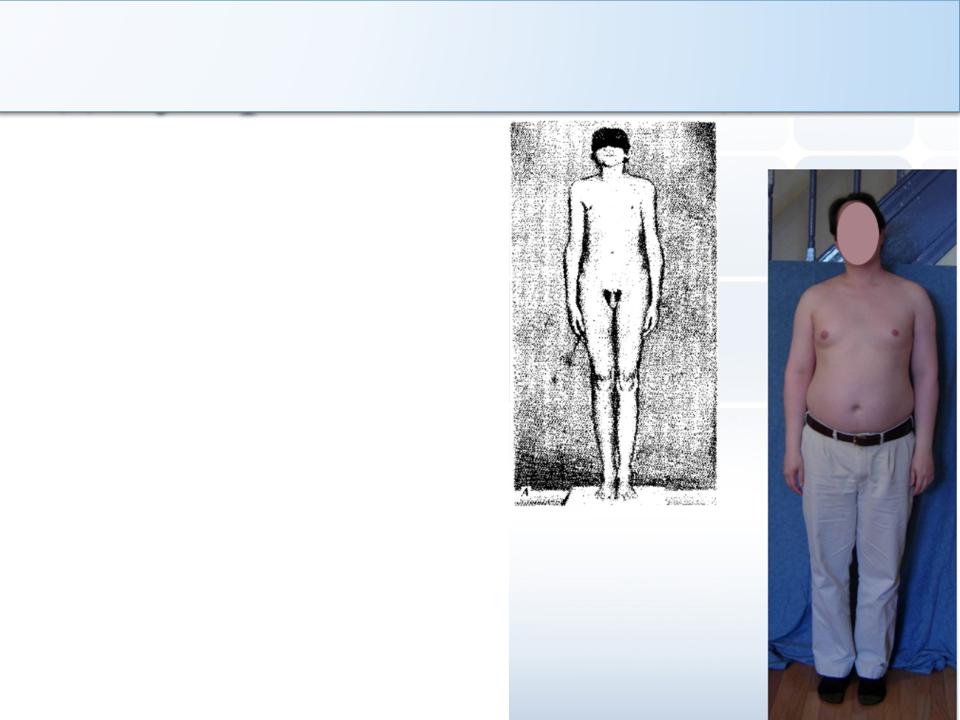 Небольшая часть тестостерона циркулирует в крови в свободной форме в динамическом равновесии со связанными фракциями [3].
Небольшая часть тестостерона циркулирует в крови в свободной форме в динамическом равновесии со связанными фракциями [3].
Основная часть циркулирующего в плазме крови тестостерона преобразуется в более активную форму гормона – в дигидротестостерон (ДГТ) под воздействием фермента 5a–редуктазы (в коже, печени, предстательной железе). Часть тестостерона метаболизируется в эстрадиол посредством фермента ароматазы. Ароматизация тестостерона происходит в яичках, головном мозге и жировых тканях. Во многих тканях активность тестостерона зависит от его восстановления до дигидротестостерона, который также связывается цитозольными андрогеновыми рецепторами, затем происходит транслокация стероид–рецепторного комплекса в ядро, где он активирует транскрипцию и изменения на клеточном уровне, связанные с действием андрогенов [5,6].
У взрослого мужчины андрогены необходимы для поддержания репродуктивных функций и вторичных половых признаков. Тестостерон также влияет на мышечную силу и массу, распределение жировой ткани, костную массу, эритропоэз, сперматогенез, а также на половое влечение и потенцию. Кроме того, андрогены могут оказывать влияние на общий метаболизм, настроение и самочувствие.
Таким образом, андрогены учавствуют в поддержании здоровья мужчины в зрелые годы. Несмотря на то, что дефицит андрогенов оказывает отрицательное воздействие на организм мужчины, диагностике и лечению мужского гипогонадизма обычно уделяется недостаточно внимания. Пациенты часто отрицают наличие у них симптомов дефицита тестостерона, приписывая их другим заболеваниям, и клиницистам не во всех случаях удается их распознавать. По мере накопления информации о важной роли андрогенов для функционирования и здоровья мужского организма, мужской гипогонадизм становится предметом все большего внимания. В последние годы появляется все больше научных исследований, свидетельствующих в пользу применения заместительной терапии тестостероном у взрослых мужчин.
Основной целью заместительной терапии тестостероном является достижение концентрации тестостерона в крови, максимально приближенной к физиологическим значениям у мужчин с нормальной функцией яичек [7]. В качестве этой терапии должны использоваться физиологические дозы тестостерона, а уровни тестостерона, дигидротестостерона и эстрадиола поддерживаться в пределах нормальных значений. Кроме того, препараты должны быть безопасны, а лекарственная форма удобна для пациента. В ряде исследований было показано, что применение заместительной терапии тестостероном у мужчин с гипогонадизмом приводит к восстановлению массы тела и снижению объема жировой ткани [8]. При этом отмечается повышение либидо, улучшение сексуальной функции, а также самочувствия и настроения [5,8,9].
В качестве этой терапии должны использоваться физиологические дозы тестостерона, а уровни тестостерона, дигидротестостерона и эстрадиола поддерживаться в пределах нормальных значений. Кроме того, препараты должны быть безопасны, а лекарственная форма удобна для пациента. В ряде исследований было показано, что применение заместительной терапии тестостероном у мужчин с гипогонадизмом приводит к восстановлению массы тела и снижению объема жировой ткани [8]. При этом отмечается повышение либидо, улучшение сексуальной функции, а также самочувствия и настроения [5,8,9].
Лечение, направленное на нормализацию уровня тестостерона, представляется патогенетической терапией гипогонадизма, особенно у пожилых мужчин с возрастным андрогенным дефицитом.
При отсутствии лечения гипогонадизм может быть фактором риска развития остеопороза у мужчин, а применение заместительной гормональной терапии повышает плотность костной ткани. Андрогены стимулируют выработку эритроцитов путем увеличения образования эритропоэтина. Экзогенные андрогены вызывают подавление эндогенного тестостерона по механизму отрицательной обратной связи через снижение выработки ЛГ. Большие дозы андрогенов могут привести к подавлению и сперматогенеза в результате ингибирования продукции ФСГ.
Андрогены стимулируют синтез белка и подавляют их катаболизм, т.е обладают анаболическим эффектом.
Арсенал средств для заместительной терапии андрогенами постоянно расширяется, и в последнее время для лечения гипогонадизма применяются производные андрогенов для перорального применения, внутримышечное введение тестостерона длительного действия, трансдермальный пластырь с тестостероном.
Для перорального или сублингвального приема используются некоторые 17a–алкильные производные тестостерона (например, метилтестостерон, флуоксиместерон, оксандролон). Все эти препараты метаболизируются в печени, их необходимо принимать несколько раз в день, их андрогенные свойства низкие или варьируют. Более того, они гепатотоксичны и из–за особенностей метаболизма могут повышать уровень липопротеидов низкой плотности и резко снижать содержание липопротеидов высокой плотности.
Для внутримышечного введения применяются инъекции депо эфиров тестостерона (тестостерона энантат и тестостерона ципионат) в масляной суспензии. Иньекции могут назначаться 1 раз в 2–3 недели в обычной дозировке для взрослых 150–200 мг [5]. В первые несколько дней эти препараты повышают уровень тестостерона до верхней границы нормы или несколько выше, который затем снижается до нижней границы нормы или ниже в конце интервала между введениями. Такие колебания уровня тестостерона в широких пределах могут привести к колебаниям либидо и настроения и могут привести к появлению акне, полицитемии и развитию гинекомастии.
Трансдермальный пластырь обеспечивает медленное поступление тестостерона в кровоток, не требует применения производных тестостерона. Эти системы обеспечивают физиологический уровень тестостерона в плазме, имитируя суточный ритм, наблюдаемый у здоровых мужчин. При этом не наблюдается колебаний, которые имеют место при применении иньекционных форм. Однако многие мужчины считают трансдермальный путь поступления тестостерона неприемлемым с косметической точки зрения. Кроме того, пластыри могут вызвать раздражение кожи, могут отклеиваться. Эта форма доставки тестостерона неудобна также из–за размера пластыря и необходимости еженедельной подготовки кожи мошонки. Приклеивание пластыря на мошонку приводит к некоторому повышению уровня дигидротестостерона, по–видимому, из–за высокого уровня 5a–редуктазы в коже мошонки, что приводит к усилению метаболизма тестостерона.
Прикрепление пластыря на других частях тела часто сопровождается местными кожными реакциями и тем самым приводит к прерыванию лечения.
В последние годы большое внимание уделяется исследованию преимуществ трансдермальных препаратов тестостерона.
Наиболее приемлемым является применение тестостерона в виде трансдермального геля, который наносится один раз в день на кожу (обычно в области плеча, надплечья или живота). Местные реакции при применении геля минимальны. Нанесение геля на кожу неинвазивно, безболезненно и может производиться в домашних условиях. Нанесенный гель незаметен для окружающих. Тестостерон в виде геля всасывается с поверхности кожи и поступает в кровоток, при этом постоянная концентрация тестостерона в крови поддерживается в течение 24 часов. В течение трех дней терапии пониженный уровень тестостерона возвращается к норме [9,10].
Нанесенный гель незаметен для окружающих. Тестостерон в виде геля всасывается с поверхности кожи и поступает в кровоток, при этом постоянная концентрация тестостерона в крови поддерживается в течение 24 часов. В течение трех дней терапии пониженный уровень тестостерона возвращается к норме [9,10].
Не рекомендуется нанесение геля на область половых органов. Следует избегать контакта кожи больного с кожей другого человека.
Тестостерон при нанесении на поверхность кожи быстро высыхает и всасывается в кожу, в результате создается резервуар, из которого происходит его постоянное поступление в кровоток в течение 24 часов. При однократном применении – приводит к достижению нормального уровня тестостерона в крови через 1–2 часа после нанесения, и этот уровень поддерживается в течение 24 часов. После прекращения применения средняя концентрация тестостерона в крови оставалась нормальной в течение 24–48 часов, а затем снижалась до исходного уровня на 4–й день после последнего нанесения. Исследования [10] показали, что при достижении нормальных значений тестостерона и продолжении приема препарата в той же дозировке уровень тестостерона в плазме крови поддерживается в пределах нормальных значений на 30, 90 и 180–й день после начала терапии. Было показано, что повышение уровня тестостерона зависит от дозы применяемого препарата, и каждые 2,5 г нанесенного препарата приводят к повышению средней суточной концентрации тестостерона на 125 нг/дл [11].
Около 2% циркулирующего тестостерона остается свободным (несвязанным), 44% связывается с секс–стероидсвязывающим глобулином (СССГ) и 54% слабо связывается с альбумином и другими белками [6]. Альбумин–связанная фракция не прочная, легко диссоциирует и вместе со свободной фракцией может стать биодоступной. Фракция, связанная с СССГ, не обладает биодоступностью.
Инактивация тестостерона происходит главным образом в печени. Наиболее активными метаболитами тестостерона являются эстрадиол и ДГТ. В течение 30 дней от начала терапии тестостероном уровень эстрадиола в крови значительно возрастает и остается повышенным в течение всего периода лечения, но не выходит за пределы величин, характерных для мужчин с сохраненной функцией яичек.
В период лечения гелем тестостерона возрастает концентрация ДГТ параллельно с увеличением уровня тестостерона. Проведенные исследования [11] показали, что средняя концентрация ДГТ в течение 180 дней терапии остается в пределах нормальных значений.
Перед назначением лечения должны быть учтены не только показания, но и противопоказания к применению препарата.
Показания: первичный и вторичный гипогонадизм у мужчин.
Противопоказания:
• Андрогены противопоказаны мужчинам при наличии карциномы или рака грудной железы и при подозрении на их наличие.
• Тестостерон не показан женщинам. Беременные женщины должны избегать любого контакта препарата с кожей. В случае контакта контактировавший участок кожи должен быть немедленно вымыт водой с мылом.
• Тестостерон не должен использоваться пациентами с гиперчувствительностью.
• Продолжительное применение высоких доз тестостерона может привести к побочным эффектам со стороны печени.
• У пожилых больных под воздействием андрогенов может повышаться риск прогрессии субклинического рака предстательной железы. Такие пациенты перед началом заместительной терапии тестостероном должны быть обследованы на предмет выявления этого заболевания.
• У пациентов, получающих терапию по поводу гипогонадизма, часто встречается гинекомастия.
• У пациентов, страдающих тяжелой сердечной, почечной или печеночной недостаточностью, лечение гелем тестостерона может вызвать осложнения, характеризующиеся отеками с застойной сердечной недостаточностью или без нее. В этом случае лечение должно быть немедленно прекращено и может потребоваться терапия диуретиками.
Под нашим наблюдением находились 17 мужчин, которые жаловались на ухудшение настроения, снижение полового влечения и либидо, быструю утомляемость, снижение мышечной силы. У 7 больных с гипогонадизмом не было выявлено никаких изменений, у 3 пациентов была обнаружена умеренная гинекомастия, уменьшение количества волос на лице и теле. Яички пальпаторно были мягкими и небольших размеров.
У 4 обследованных отмечалось ожирение (индекс массы тела составлял 32±0,03 кг/м2). У 9 пациентов отмечалось абдоминальное ожирение (110±4 см). Возраст пациентов колебался от 18 до 58 лет. Всем пациентам проводили УЗИ гонад и исследование уровня тестостерона, эстрадиола, ЛГ и ФСГ в плазме крови, а также содержание СССГ.
Как указывалось выше, большинство форм гипогонадизма у взрослых мужчин являются приобретенными и часто развиваются на фоне ожирения, тяжелых системных заболеваний, приема некоторых лекарств, серповидно–клеточной анемии, СПИДа, цирроза печени и почечной недостаточности. Все указанные причины были исключены до начала лечения.
По результатам обследования первичный гипогонадизм был диагностирован у 5 пациентов и вторичный соответственно у 12.
Лечение больных проводили водно–спиртовым гелем, содержащим 1% тестостерона, который наносили на чистую, сухую, неповрежденную кожу в области плеча, надплечья или живота 1 раз в сутки. Препарат обеспечивает поступление тестостерона в течение 24 часов. 5 г андрогеля содержат 50 мг тестостерона, примерно 10% дозы тестостерона, ежедневно наносимого на поверхность кожи, поступает в системный кровоток. Препарат обеспечивает поступление тестостерона в физиологических количествах и поддерживает уровень циркулирующего тестостерона в пределах нормы, как у здоровых мужчин.
Применение препарата продолжали от 3 до 4 месяцев. При необходимости пациентам назначали курс поддерживающей терапии: витаминами, применяли седативные средства и адаптогены.
При исследовании уровня тестостерона через 30 дней после начала лечения констатировали нормальное его содержание, при этом у пациентов с первичным гипогонадизмом концентрация ЛГ и ФСГ снижалась, но не достигала нормального уровня, и только через 2 месяца после начала терапии уровень обоих гормонов достигал нормальных величин. У больных с вторичным гипогонадизмом нормальные или сниженные концентрации ЛГ и ФСГ через 30 и 90 дней после начала терапии снижались до нижней границы нормы или несколько ниже.
Содержание эстрадиола в плазме крови повышалось значительно уже в первые 30 дней от начала лечения и оставалось повышенным в течение всего периода исследования (2–3 месяца), но не выходило за пределы нормальных значений для мужчин с сохраненной функцией яичек.
Средний уровень СССГ в течение 3 месяцев исследования практически не изменился ни в одной из групп.
Заместительная терапия гелем тестостерона в течение 3–4 месяцев благоприятно отразилась на состоянии пациентов. У подавляющего большинства из них (у 14) отмечалось значительное улучшение настроения, повышение работоспособности и физической активности, либидо, сексуальной активности. Эти улучшения начались уже в первые 2 недели от начала приема препарата и продолжались в течение всего периода лечения. У 3 пациентов с возрастным гипогонадизмом улучшение настроения, общего самочувствия, работоспособности появились через 2 месяца после начала приема препарата.
Применение заместительной гормональной терапии тестостероном привело к прогрессивному снижению массы жировой ткани у всех пациентов, что проявлялось уменьшением окружности талии (в среднем на 4–5 см за 1 месяц) и повышению мышечной массы в обоих группах пациентов.
При нанесении геля тестостерона на кожу лишь у двух пациентов наблюдалась местная реакция в виде легкого зуда кожи, который был краткосрочным и не потребовал прекращения применения препарата или лечения. Из других побочных эффектов: у одного больного появились головные боли на фоне приема препарата, в одном случае имела место астения и повышение артериального давления. В проведенном исследовании заболеваний предстательной железы или яичка мы не констатировали.
В проведенном нами исследовании применение 5 г препарата не оказывало отрицательного действия на функциональное состояние печени, в то же время пероральные препараты тестостерона при длительном применении обладают выраженной гепатотоксичностью (токсический гепатит, опухоли печени, холестатический гепатит и желтуха) [9].
При местном применении андрогенов возможна передача препарата через кожу при контакте с партнершой. Для предупреждения риска передачи андрогенов партнерше и ее вирилизации место нанесения препарата рекомендуется прикрыть одеждой. Исследования [9] показали, что при прикрытии места нанесения препарата одеждой передача тестостерона партнерше пациента полностью предотвращалась. Если возникла ситуация, при которой контакт с кожей других лиц неизбежен, пациент должен принять душ. Для оптимального всасывания тестостерона достаточно 5–6 часов, после чего пациентам рекомендуется принимать водные процедуры.
У пациентов, длительное время получающих заместительную терапию андрогенами, следует периодически исследовать уровни гемоглобина и гематокрита, а также холестерина и липопротеидов высокой плотности.
Необходимо исследовать функцию печени и определять величину простат–специфического антигена.
Для правильного подбора дозы андрогенов необходимо исследовать уровень тестостерона в плазме крови.
Таким образом, применение препаратов тестостерона в качестве заместительной терапии у больных с гипогонадизмом патогенетически обосновано и оказывает позитивное влияние на все звенья полового акта (оргазм, эрекция, половое влечение, эякуляция). Препараты, содержащие 1%–й тестостерон, обладают минимальными побочными действиями и могут быть рекомендованы для терапии первичного и вторичного гипогонадизма.
Литература
1. В кн.: Болезни эндокринной системы – М:Медицина, 2000.–С.520–528.
2. Wilson J.D.Androgens.In:Gilman A.G.,consulting ed; Hardman J.G., Limbird L.E.,eds–in chief, Molinoff P.B., Ruddon R.W., eds.Goodman and Gillmans/The Pharmacologic Basis of Therapeutics.8th ed.New York, NY : Pergamon Press, Inc.,1990.– P.1413–1430.
3. Bagatell C.J., Bremner W.J. Drug therapy: androgens in men – uses and abuses.–
N.Engl.J.Med..–1996; 334.– P.707–714.
4. Tenover J.L. Male hormone replacement therapy including *andropause*. Endocrinol.
Endocrinol.
Metab.Clin.North Am.– 1998;27.–P.969–987.
5. Winters S.J. Current status of testosterone replacement therapy in man. Arch. Fam.Med.–
1999.–8.– P.257–263.
6. Griffin J.E., Wilson J.D. Disorders of the testes and the male reproductive tract. In : Wilson J.D., Foster D.W., Kronenberg H.M., Larsen P.R.,eds. Williams Textbook of
Endocrinology.9th ed.Philadelphia, Pa:W.B. Saunders Co;1998.– P.819–875.
7. Nieschlag E., Behre H.M. Pharmacology and clinical uses of testosterone. In: Nieschlag E., Behre H.M., eds. Testosterone: Action, Deficiency, Substitution. 2nd ed.Berlin – Heidelberg: Springer–Verlag: 1998.– P.293–328.
8. Wang C., Swerdloff R.S. Androgen replacement therapy. Ann.Med. 1997.–29.–P. 365–370.
9. Wang C., Berman N. Longstreth J.A. et al. Pharnmacokinetics of transdermal testosterone gel in hypogonadal men: application of gel at one site versus four sites: a general research center study./ J.Clin.Endocrinol.Metab.–2000.–85.– P.964–966.
10. Wang C., Swerdloff R.S., Iranmanesh A. et al.Transdermal testosterone gel improves sexual function, mood, muscle strength, and body composition parameters in hypogonadal men. J.Clin. Endocrinol. Metab.–2000.–85.– P.2839–2853.
11. Swerdloff R.S., Wang C., Cunningham G. et al. Long– term pharmacokinetics of transdermal gel in hypogonadal men./ J.Clin. Endocrinol. Metab.–2000.–85.– P.4500–4510.
.
Синдром гипогонадизма у мужчин | Rozhivanov
1. Андрология. Мужское здоровье и дисфункция репродуктивной системы Под ред. Э. Нишлага, Г.М. Бере. М., 2005. 554 с.
2. Дедов И.И., Семичева Т.В., Петеркова В.А. Половое развитие детей: норма и патология. М., 2002. 232 с.
3. Роживанов Р.В., Шурдумова Б.О., Парфенова Н.С., Савельева Л.В. Комплексный подход к лечению ожирения и метаболического синдрома у мужчин. Ожирение и метаболизм. 2009;4:38-41.
Роживанов Р.В., Шурдумова Б.О., Парфенова Н.С., Савельева Л.В. Комплексный подход к лечению ожирения и метаболического синдрома у мужчин. Ожирение и метаболизм. 2009;4:38-41.
4. Роживанов Р.В. Эндокринные нарушения половой функции у мужчин / в. кн. Рациональная фармакотерапия заболеваний эндокринной системы и нарушений обмена веществ. 2-е издание. под. ред. Дедова И. И., Мельниченко Г.А. 2013:754-770.
5. Савельева Л.В., Роживанов Р.В., Шурдумова Б.О., Фадеев В.В. Нормогонадотропный гипогонадизм у мужчин с ожирением. Ожирение и метаболизм. 2009;3(20):39-42.
6. Heufelder А, Gooren L, Bunck M, Saad F. Testosterone Treatment Enhances the Favorable Effects of Exercise and Diet on Inflammation, Metabolism and Coagulation Markers in Hypogonadal Men with the Metabolic Syndrome. Presented at The Endocrine Society Annual Meeting, Toronto, Canada, June 2-5, 2007. ENDO Abstracts 2007: OR35-2.
7. Heufelder А, Gooren L, Bunck M, Saad F. Treatment with Diet and Exercise Plus Transdermal Testosterone Reverses the Metabolic Syndrome and Improves Glycemic Control in Hypogonadal Men with Newly Diagnosed Type 2 Diabetes. Presented at The Endocrine Society Annual Meeting, Toronto, Canada, June 2-5, 2007. ENDO Abstracts 2007:P2-272.
8. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. // J Clin Endocrinol Metab. 2004 Jun;89(6):2548-56.
9. Morales A, Lunenfeld B. Investigation, treatment and monitoring of late-onset hypogonadism in males. The Aging Male. 2002;5:74-86.
10. Svartberg J et al. Waist circumference and testosterone levels in community dwelling men. The Tromsø study. Europ J Epidemiol. 2004;19(7):657-63.
Svartberg J et al. Waist circumference and testosterone levels in community dwelling men. The Tromsø study. Europ J Epidemiol. 2004;19(7):657-63.
11. Tsai EC, Boyko EJ, Leonetti DL, Fujimoto WY. Low serum testosterone level as predictor of increased visceral fat in Japanese-American men. Int J Obes Relat Metab Disord. 2000;24:485-91.
Синдром гипогонадизма у мужчин: взгляд уролога
| Дмитрий Геннадьевич Курбатов Д.м.н., проф., руководитель отделения андрологии и урологии ФГУ «Эндокринологический научный центр Росмедтехнологий» [email protected] | |
| Роман Викторович Роживанов К.м.н., врач эндокринолог-андролог, ст. науч. сотр. отделения андрологии и урологии ФГУ «Эндокринологический научный центр Росмедтехнологий» |
С давних времен известно, что для нормальной работы мужского организма необходимо наличие сохранной функции яичек, ведь при нарушении их деятельности или удалении органа развиваются тяжелые соматические заболевания, не только ухудшающие половую функцию и снижающие качество жизни, но и сокращающие ее продолжительность. Так, у евнухов отмечается выраженное ожирение, дислипидемия, увеличивается риск развития атеросклероза, ишемической болезни сердца и многих других грозных заболеваний. Кроме того, все больше внимания уделяется урологическим проявлениям снижения функции яичек, в том числе эректильной дисфункции, бесплодию, недоразвитию наружных половых органов (микропенис), а в последние годы – и заболеваниям предстательной железы (ПЖ).
Для описания данного состояния используется множество определений: андрогенный дефицит, тестикулярная недостаточность, но наиболее удачным и принятым международными сообществами эндокринологов и урологов определением является синдром гипогонадизма у мужчин – клинический и/или лабораторный синдром, обусловленный снижением секреции половых гормонов яичками.
В зависимости от патогенеза синдром гипогонадизма у мужчин разделяют на:
- гипергонадотропный или первичный (типичным примером является анорхизм),
- гипогонадотропный или вторичный (типичный пример – синдром Каллмана или состояния, возникающие вследствие удаления опухолей гипофиза),
- нормогонадотропный (как правило, это смешанная форма гипогонадизма), типичным примером является гипогонадизм, развивающийся у мужчин с ожирением или по мере увеличения возраста.
Данные по частоте встречаемости как первичного, так и вторичного гипогонадизма у мужчин базируются на распространенности основных причин его развития. Так, анорхизм встречается у 3–5% мальчиков с отсутствием яичек в мошонке. Синдром Клайнфельтера (генетическое заболевание, для которого характерен первичный гипогонадизм) встречается у 1 из 500 мальчиков, а синдром Каллмана (заболевание гипофиза, при котором развивается вторичный гипогонадизм) – у 1 из 5000. Распространенность возрастного гипогонадизма колеблется в пределах 10–50% в зависимости от возрастной группы. В Массачусетском исследовании пожилых мужчин (1994) было установлено, что уровень биодоступного тестостерона начинает снижаться уже с 30–35 лет со скоростью около 2–3% в год, а общего – с 50–55 лет на 0,8–1,6% в год. Схожие изменения получены и в других исследованиях. Распространенность гипогонадизма при ожирении точно не установлена, но по данным ФГУ «Эндокринологический научный центр» она достигает 30–50%.
Гипергонадотропный гипогонадизм обусловлен снижением или полным отсутствием андрогенсекретирующей функции яичек, вследствие их поражения патологическим процессом, а гипогонадотропный гипогонадизм – снижением или полным выпадением гонадотропной стимуляции яичек. При возрастном гипогонадизме происходит снижение продукции тестостерона яичками вследствие ухудшения их функции, апоптоза клеток Лейдига, ишемических изменений паренхимы яичек и, возникающих на фоне увеличения возраста, гипоталамо-гипофизарных нарушений.
В зависимости от времени возникновения выделяют: препубертатный и постпубертатный гипогонадизм. Следует отметить, что для препубертатного гипогонадизма характерны высокий рост, евнухоидные пропорции тела, распределение жира по женскому типу, гинекомастия, отсутствие оволосения, высокий тембр голоса и недоразвитие половых органов (микропенис). Для постпубертатного гипогонадизма характерно снижение половой функции и тургора яичек, развитие соматических синдромов.
К развитию первичного гипогонадизма приводят: синдром Клайнфельтера, анорхизм, травмы, облучение, химиотерапия по поводу рака, а также другие токсические поражения яичек, поздно леченый крипторхизм. К развитию вторичного – синдромы Каллмана (дефицит ЛГ и ФСГ в сочетании с аносмией), «фертильного евнуха» (изолированный дефицит ЛГ), опухоли гипофиза и гипоталамуса, их хирургическое лечение или лучевая терапия, кровоизлияния в опухоли.
Учитывая, что биологически активной является фракция свободного тестостерона, полноценная оценка андрогенного статуса пациента, особенно в спорных ситуациях, должна включать в себя не только определение в крови уровней ЛГ, ФСГ и общего тестостерона, но и глобулина, связывающего половые гормоны, или свободного тестостерона.
Диагноз заболевания и его конкретной формы устанавливают на основании характерной клинической картины, подтвержденной данными лабораторного обследования (схема 1).
Схема 1. Диагностический алгоритм синдрома гипогонадизма
Основная симптоматика гипогонадизма хорошо известна и подробно описана в литературе. Необходимо лишь подчеркнуть, что, к сожалению, нет какого-либо одного-двух строго специфических симптомов данного состояния, поэтому клиническая картина часто вовремя не распознается или замаскировывается другими соматическими проявлениями. К урологам больные чаще обращаются с жалобами сексуального характера. При этом известно, что эректильная дисфункция (ЭД) и снижение либидо могут быть единственным проявлением гипогонадизма, а при скрининговых обследованиях дефицит тестостерона выявляется у 18,3–30% больных с ЭД (Schulman С. и соавт., 2006; Yassin и соавт., 2006; Дедов И.И., 2006).
и соавт., 2006; Yassin и соавт., 2006; Дедов И.И., 2006).
Основными задачами терапии синдрома гипогонадизма у мужчин являются: полное устранение дефицита тестостерона – восстановление или развитие вторичных половых признаков, либидо и эрекции, повышение мышечной силы, лечение или профилактика остеопороза, устранение сопутствующих соматических синдромов, депрессии, восстановление фертильности.
Согласно рекомендациям ISA, ISSAM, EAU, EAA и ASA по выявлению, лечению и мониторированию гипогонадизма у мужчин (2008), его устранение достигается назначением терапии препаратами тестостерона. Основными различиями между препаратами служат особенности их фармакокинетики, а также спектр фармакодинамической активности.
На сегодняшний день в России представлены следующие препараты: пероральная и инъекционная форма тестостерона ундеканоата, смеси эфиров тестостерона для парентеральной терапии и гелевая форма тестостерона. Ранее широко использовавшийся метилтестостерон, который является алкилированным препаратом тестостерона, разработанным для пероральной терапии, в настоящее время не применяется в связи с высокой гепатотоксичностью. Тестостерона ундеканоат для перорального приема лишен гепатотоксичности и в терапевтических дозах не угнетает эндокринную функцию яичек и сперматогенез. Однако недостатками препарата служат его относительно слабое андрогенное действие, а так же многократность приема. Препараты смесей эфиров тестостерона для парентеральной терапии являются наиболее распространенными, так как их состав позволяет добиться быстрого и продолжительного эффекта. Обычная схема их применения – по 1 мл внутримышечно 1 раз в 3 нед. Недостаток этих препаратов заключается в возникновении супрафизиологических пиков концентрации тестостерона в первые несколько дней после инъекции. Препарат тестостерона ундеканоат для парентерального введения не дает подобных пиков концентрации тестостерона и применяется путем внутримышечных инъекций 1 раз в 3 мес. Гель, содержащий тестостерон, наносится непосредственно на кожу ежедневно.
Для устранения гипогонадизма (за исключением гипергонадотропного) при необходимости репродуктивной реабилитации возможно так же применение терапии препаратами гонадотропинов. Для этих целей используются препараты хорионического гонадотропина, лютеинизирующего и фолликулостимулирующего гормонов, которые назначаются курсом на период до 3–6 мес. Дозу препаратов выбирают исходя из результатов серийных определений уровня тестостерона в сыворотке крови и контрольных исследований спермограммы. Мужчины с исходным объемом яичек более чем 4 см3 имеют лучший прогноз для восстановления фертильности.
Наряду с препаратами тестостерона и гонадотропинов потенциальными кандидатами на роль средств для устранения гипогонадизма являются антиэстрогены, а также препараты дигидротестостерона и дегидроэпиандростерона, однако к настоящему времени проведено недостаточное число клинических исследований.
На фоне терапии у больных с гипогонадизмом устраняются основные проявления данного состояния, развиваются и поддерживаются вторичные половые признаки. Доказано, что терапия тестостероном эффективно восстанавливает эректильную функцию у большинства пациентов с ЭД, обусловленной гипогонадизмом, при этом лечение тестостероном может рассматриваться как терапия 1-й линии. Эффективность лечения обусловлена тем, что тестостерон оказывает комплексное действие: нормализует функцию эндотелия и стимулирует секрецию оксида азота, улучшает артериальный приток к кавернозным артериям, усиливает сократимость бульбокавернозных мышц и увеличивает интракавернозное давление. Кроме того, по нашим данным, а также данным некоторых зарубежных исследователей было доказано, что применение тестостерона восстанавливает веноокклюзионную функцию у мужчин с гипогонадизмом и веногенной ЭД.
Что касается ПЖ, основного «фактора страха» для специалистов, то в настоящее время отсутствуют данные о том, что терапия андрогенами стимулирует развитие доброкачественной гиперплазии или рака ПЖ (РПЖ) (Лоран О. Б., Сегал А.С., 1999; Hermann M., Berger P., 1999; Morales A., Lunenfeld B., 2002; Dean J.D. и соавт., 2004; Rhoden E.L., Morgentaler A., 2005; Wang C. и соавт., 2007). Так, установлено, что распространенность, как доброкачественной гиперплазии, так и РПЖ увеличивается с возрастом, при этом уровень тестостерона снижается. Отсутствует связь между объемом ПЖ и уровнем тестостерона в крови, как у эугонадных мужчин, так и при гипогонадизме.
Б., Сегал А.С., 1999; Hermann M., Berger P., 1999; Morales A., Lunenfeld B., 2002; Dean J.D. и соавт., 2004; Rhoden E.L., Morgentaler A., 2005; Wang C. и соавт., 2007). Так, установлено, что распространенность, как доброкачественной гиперплазии, так и РПЖ увеличивается с возрастом, при этом уровень тестостерона снижается. Отсутствует связь между объемом ПЖ и уровнем тестостерона в крови, как у эугонадных мужчин, так и при гипогонадизме.
В работе, проведенной группой по изучению эндогенных половых гормонов и РПЖ (2008), были проанализированы результаты 18 проспективных исследований у 3886 больных с РПЖ и 6438 здоровых мужчин.
Были исследованы уровни большинства половых гормонов (тестостерона, дигидротестостерона, дегидроэпиандростерона сульфата, андростендиона и эстрадиола), что позволило прийти к выводу об отсутствии связи этих гормонов с риском развития рака.
Заместительная терапия андрогенами не влияет на объем ПЖ и уровень простатического специфического антигена (ПСА) в крови, и не ухудшает максимальную скорость потока мочи (Роживанов Р.В., 2010). Так же установлено, что терапия тестостероном не увеличивает риск развития рака в железе, в связи с повышением индекса апоптоза и атрофии и снижением соотношения эпителиальных/ стромальных клеток в периферических зонах ПЖ, основных зонах развития злокачественного процесса (Efesoy O., 2010).
Однако низкий уровень тестостерона в крови ассоциируется с более злокачественным течением и худшим прогнозом РПЖ (Schatzl G. и соавт., 2000; Liu C.C., 2006), и взаимосвязан с подтвержденным диагнозом РПЖ (GarciaCruz E., 2010). Мужчины с уровнем тестостерона ниже 200 нг/дл (7 нмоль/л) в 3 раза чаще умирают от РПЖ (Massengill J.C., 2003; Kratzik C., 2006), нежели чем пациенты без гипогонадизма. Наличие манифестного РПЖ является абсолютным противопоказанием к назначению терапии андрогенами, кроме того, тестостерон может стимулировать прогрессирование субклинического РПЖ (Hermann M., Berger P., 1999; Morales A. , Lunenfeld B., 2002; Rhoden E.L., Morgentaler A., 2004; Lunglmayr G., 2006; Лоран О.Б. и соавт., 2007). Поэтому больные, получающие терапию андрогенами, должны подвергаться периодическому мониторингу: клиническим осмотрам и лабораторным тестам. Частота периодического наблюдения зависит от возраста больного. У молодых мужчин эти тесты могут выполняться с ежегодными интервалами, у пожилых – каждые 3–6 мес. Мониторированию подлежит концентрация тестостерона, гемоглобина, гематокрита и уровень ПСА у мужчин старше 40 лет, а так же пальцевое ректальное обследование ПЖ. Следует отметить, что уровень ПСА может быть ниже при гипогонадизме, чем у здоровых мужчин, и терапия тестостероном может повышать его уровень, но не до уровня эугонадных мужчин. Рост ПСА не более 0,5 нг/мл в год является безопасным и не требует отмены терапии.
, Lunenfeld B., 2002; Rhoden E.L., Morgentaler A., 2004; Lunglmayr G., 2006; Лоран О.Б. и соавт., 2007). Поэтому больные, получающие терапию андрогенами, должны подвергаться периодическому мониторингу: клиническим осмотрам и лабораторным тестам. Частота периодического наблюдения зависит от возраста больного. У молодых мужчин эти тесты могут выполняться с ежегодными интервалами, у пожилых – каждые 3–6 мес. Мониторированию подлежит концентрация тестостерона, гемоглобина, гематокрита и уровень ПСА у мужчин старше 40 лет, а так же пальцевое ректальное обследование ПЖ. Следует отметить, что уровень ПСА может быть ниже при гипогонадизме, чем у здоровых мужчин, и терапия тестостероном может повышать его уровень, но не до уровня эугонадных мужчин. Рост ПСА не более 0,5 нг/мл в год является безопасным и не требует отмены терапии.
С другой стороны, было установлено, что, используемая в лечении распространенного РПЖ, постоянная андрогенная депривация сопровождается манифестацией сахарного диабета или метаболического синдрома и повышает смертность от сердечно-сосудистых заболеваний (снижение уровня тестостерона на 6 нмоль/л повышает смертность на 14%). Интермиттирующая андрогенная супрессия идентична по выживаемости, но превосходит по улучшению качества жизни больных, т. е. является возможной альтернативой постоянной андрогенной блокаде (Maggi и соавт., 2007; Kwah и соавт., 2007; Conti и соавт., Cochrane Database Syst Rev., 2007).
В последнее время были предприняты исследования, направленные на изучение возможности применения андрогенов после радикального лечения РПЖ. Так, O. Nabulsi и соавт. (2008), назначая заместительную терапию гелевой формой тестостерона в течение 2 лет 22 мужчинам с гипогонадизмом, перенесшим радикальную простатэктомию, отметили возникновение лабораторных признаков рецидива (повышение ПСА) только у 1 пациента через 17 мес после операции. Это позволило исследователям сделать вывод о возможности проведения такой терапии под строгим медицинским контролем.
Следует отметить, что в последние годы накапливается все больше данных об эффективности и безопасности терапии гипогонадизма у пациентов с другими урологическими заболеваниями и синдромами. Так, примерно у 20% пожилых мужчин с симптомами нижних мочевых путей (СНМП) имеются проявления гипогонадизма, при этом симптомы могут быть его первичным проявлением (Schatzl G. и соавт., 2000). По данным И.А. Корнеева и С.Ю. Глазневой (2008) у 78 мужчин (средний возраст 57,7 лет) с возрастным гипогонадизмом были обнаружены более выраженные жалобы на расстройства мочеиспускания, чем в контроле.
Развитие гипогонадизма усугубляет тяжесть состояния у больных с синдромом хронической тазовой боли (СХТБ), для которых, по данным Д.Г. Курбатова и соавт. (2007), в сравнении с эугонадными пациентами характерны: более выраженный болевой синдром, низкое качество сексуальной функции, повышенный уровень тревожности, депрессии и астении. Доказанные анаболическое и антиишемическое действие андрогенов, как и остальные положительные эффекты, позволяют с успехом их использовать при лечении обсуждаемых состояний (СХТБ и СНМП). Кроме того, у пациентов достоверно улучшаются настроение и общее самочувствие.
К сожалению, по доступным статистическим данным, необходимое лечение получают лишь 10–20% больных гипогонадизмом из общей популяции, что обусловлено низкой осведомленностью пациентов и врачей. Кроме того, симптомы заболевания часто не различаются, игнорируются или относятся к возрастным изменениям, а методы диагностики и цели лечения недостаточно понимаются специалистами. Так же весьма часто встречается факт «гормонофобии» не только среди урологов, но даже среди эндокринологов.
В отделении андрологии и урологии ФГУ «Эндокринологический научный центр» МЗ и СР РФ, первом в России уроандрологическом отделении, открытом на базе эндокринологического учреждения, накоплен огромный опыт безопасного лечения мужчин с синдромом гипогонадизма, что позволяет быстро и эффективно устранить любые его проявления, и, как следствие, улучшить качество и увеличить продолжительность жизни пациентов.
Средняя оценка:
Ваша оценка: Нет Средняя оценка: 5 (1 vote)
ГИПОГОНАДИЗМ — Ферти-Лайн
Гипогонадизм – синдром, сопровождающийся недостаточностью функций половых желез и нарушением синтеза половых гормонов. Гипогонадизм, как правило, сопровождается недоразвитием наружных или внутренних половых органов, вторичных половых признаков, расстройством жирового и белкового обмена (ожирением или малым весом, изменениями костной системы, сердечно-сосудистыми нарушениями). Диагностика и лечение гипогонадизма осуществляется совместно эндокринологами, гинекологами, сексопатологами и андрологами (у мужчин). Основу лечения гипогонадизма составляет заместительная гормональная терапия. При необходимости проводится хирургическая коррекция.
Женский гипогонадизм характеризуется недоразвитием и гипофункцией половых желез — яичников. Первичный гипогонадизм обусловлен либо врожденным недоразвитием яичников, либо повреждением их в период новорожденности. В организме возникает дефицит женских половых гормонов, что вызывает увеличение продукции гонадотропинов, стимулирующих яичники в гипофизе. В сыворотке крови отмечается высокий уровень фолликулостимулирующего и лютеинизирующего гормонов (гипергонадотропный гипогонадизм) и при этом низкая концентрация эстрогенов. Дефицит эстрогенов вызывает недоразвитие и атрофические изменения женских половых органов, молочных желез, первичную аменорею. Если нарушение в яичниках возникло в допубертатный период, то вторичные половые признаки отсутствуют.
Причинами первичного гипергонадотропного гипогонадизма являются врожденное генетическое нарушение (синдром Шерешевского-Тернера, кариотип 45,ХО), врожденная гипоплазия яичников, перенесенные инфекционные заболевания (сифилис, туберкулез, эпидемический паротит), ионизирующее излучение (лучевое, рентгеновское), оперативное удаление яичников, аутоиммунное поражение яичников (аутоиммунный оофорит) синдром тестикулярной феминизации (врожденное состояние, при котором внешний вид человека соответствует женщине при мужском генотипе).
Вторичный женский гипогонадизм (гипогонадотропный) возникает при гипоталамо-гипофизарной патологии, характеризуется дефицитом или полным прекращением синтеза и секреции гонадотропинов (ФСГ и ЛГ), регулирующих функцию яичников. Развивается вследствие воспалительных процессов в головном мозге (энцефалит, менингит, арахноидит), повреждающего действия опухолей головного мозга и сопровождается снижением стимулирующего действия гонадотропинов на функцию яичников.
Симптомы гипогонадизма у женщин.
Один из основных симптомов гипогонадизма (гипо-, нормо- и гипергонадотропного) в детородном периоде – нарушение менструального цикла и аменорея.
Недостаток женских половых гормонов ведет к недоразвитию половых признаков: гениталий, молочных желез, нарушению отложения жировой клетчатки по женскому типу, скудному оволосению. Если заболевание врожденное, или оно возникло в раннем детском возрасте, то вторичные половые признаки отсутствуют. Характерны узкий таз и плоские ягодицы. Если гипогонадизм развился в пубертатном периоде, половые признаки, которые уже успели развиться, сохраняются, но менструации прекращаются, ткани женских гениталий подвергаются атрофии.
Диагностика гипогонадизма у женщин
При гипогонадизме наблюдается заметное снижение содержания эстрогенов в крови, повышение уровня гонадотропинов (фолликулостимулирующего и лютеинизирующего гормонов). Ультразвуковое исследование выявляет матку, уменьшенную в размерах (гипоплазия матки), уменьшенные яичники. Рентгенография обнаруживает остеопороз или задержку формирования скелета.
Лечение гипогонадизма у женщин
При первичном гипогонадизме у женщин назначается медикаментозная заместительная терапия женскими половыми гормонами (препаратами эстрадиола). В случае наступления менструальноподобной реакции, назначают комбинированные препараты для ЗГТ (фемостон, цикло-прогинова, климен) содержащие два типа гормонов — эстрогены и гестагены. Заместительная гормональная терапия противопоказана при злокачественных опухолях молочных желез и половых органов, сердечно–сосудистых заболеваниях, болезнях почек, печени, тромбофлебите и др.
Заместительная гормональная терапия противопоказана при злокачественных опухолях молочных желез и половых органов, сердечно–сосудистых заболеваниях, болезнях почек, печени, тромбофлебите и др.
Прогноз для жизни при при гипогонадотропном гипогонадизме – благоприятный. Репродуктивная функция может быть восстановлена в полном объеме после проведения длительного и правильно разработанного лечебного курса. Оригинальные методики и схемы лечения были разработаны нашими врачами и используются многими специалистами, занимающимися проблемами бесплодия. Специалисты клиники «Ферти-Лайн» имеют опыт лечения более тысячи таких пациенток. После проведённого лечения родилось почти 900 здоровых малышей.
Если у Вас возникли вопросы о диагностике и лечении этого заболевания, Вы можете обратиться на прием к врачу-гинекологу клиники «Ферти-Лайн». Будьте здоровы!
Вопрос задает – Ольга, 40, Омск по теме: ЭКО
Здравствуйте. Меня зовут Ольга, 40 лет, мужу 41. Живем в городе Омск. В браке с 2015 года, детей нет. У мужа был крипторхизм с 2 сторон. В 7 лет делали операцию. Т.к. беременность не наступает, а время идет начали обследование, сдали:
1. спермограмму — количество сперматозоидов 0, через неделю повторили анализ — результат тот же. (приложен файл 1, 2).
2. Анализы на гормоны (файл 3):
ТТГ-1,95 (норма0,35-4,94),
Т4 свободный-10,27 (норма9-19,05),
Т3 свободный -3,31 (норма2,63-5,7),
ФСГ — 39,47 (норма 0,95-11,95),
ЛГ -11,92 (норма 0,57-12,07),
Пролактин — 166,47 (норма72,66-407,4),
Эстрадиол — 37 (норма 40,37-161,48),
тестостерон свободный 0,219 (норма 0,2188-0,7708),
тестостерон общий — 11,202 (норма 8,33-30,19),
глобулин связ пол гормоны — 34 (норма 16,2-68,5),
Индекс свободных андрогенов — 32,9 (норма 24,5-113,3),
Ингибин В — 13 (норма60-305).
Сделали узи — диагноз-гипергонадотропный гипогонадизм, азооспермия.
На консультации у андролога нам сказали что шансов нет, и возможно только ЭКО с донорской спермой. Более того порекомендовали досдать анализы на исключение рака яичка (исходя из результатов узи.)
Более того порекомендовали досдать анализы на исключение рака яичка (исходя из результатов узи.)
На приеме в другой клинике с этими же анализами — врач не сказал ничего конкретно, лишь назначил витамин Е и карнитон и сказал, что это долгое лечение, надо набраться терпения и ждать. А учитывая наш возраст — времени-то у нас как раз и нет.
Вот такие 2 разных мнения.
Про Вашу клинику прочитала в интернете, очень много хороших отзывов, клиника является одной из лучших в России.
Также понимая что медицина идет вперед большими шагами, появляются новые методы, новые технологии, но в глубинку доходит гораздо позднее, квалификация врачей совершенно на другом уровне — решили обратится за помощью к Вам.
Хотелось бы услышать Ваше мнение по нашей ситуации. Есть ли у нас шанс на своих детей? Что для этого нам надо сделать (какие-то еще анализы, обследования)? Сама я никаких обследований не проходила.
Как можно попасть к Вам на прием ?
Как это происходит с иногородними ( сколько времени необходимо, какие цены и прочее).
Этиопатогенетические аспекты центрального (гипогонадотропного) женского гипогонадизма | Локтионова
1. Назаренко Т.А. Стимуляция функции яичников. – Москва: МЕДпресс-информ; 2008.
2. Norton W. Fertility. // Nursing Management of Women’s Health – 2019. – P. 103–125. DOI:10.1007/978-3-030-16115- 6_6
3. Stamatiades G.A., Kaiser U.B. Gonadotropin regulation by pulsatile GnRH: signaling and gene expression. // Mol. Cell. Endocrinol. – 2018. – V.463. – P.131–141. DOI: 10.1016/j.mce.2017.10.015
4. Pettersson F., Fries H., Nillius S.J. Epidemiology of secondary amenorrhea: I.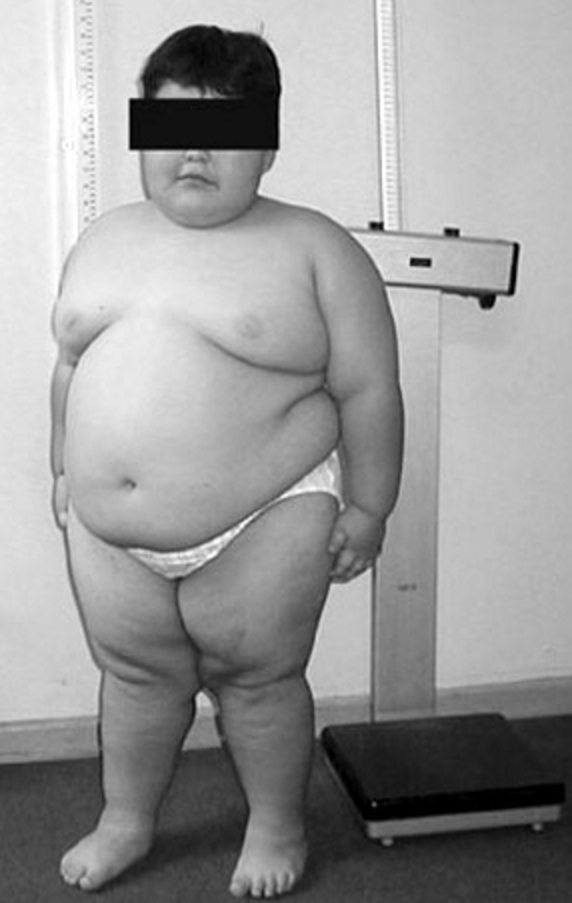 Incidence and prevalence rates. // Am. J. Obstet. Gynecol. – 1973. – V.117(1). – P. 80–86. DOI:10.1016/s0002-9378(16)33686-9
Incidence and prevalence rates. // Am. J. Obstet. Gynecol. – 1973. – V.117(1). – P. 80–86. DOI:10.1016/s0002-9378(16)33686-9
5. Meczekalski B., Katulski K., Czyzyk A., Podfigurna-Stopa A., Maciejewska-Jeske M. Functional hypothalamic amenorrhea and its influence on women’s health. // J. Endocrinol. Invest. – 2014. – V.37(11). – P.1049–1056. DOI:10.1007/s40618-014-0169-3
6. Чернуха Г.Е., Гусев Д.В., Табеева Г.И., Прилуцкая В.Ю. Патофизиологические особенности развития функциональной гипоталамической аменореи у пациенток с нервной анорексией. // Гинекология. – 2018. – Vol.20(1). DOI:10.26442/2079-5696_20.1.16-22
7. Practice Committee of the American Society for Reproductive Medicine. Current evaluation of amenorrhea // Fertility and sterility. – 2004. – V.82. – P.33-39. DOI: 10.1016/j.fertnstert.2006.08.013
8. Иловайская И.А., Зекцер В.Ю., Михайлова Д.С., Донина Е.Ю., Гончаров Н.П., Мельниченко Г.А. Функциональное состояние гипоталамо-гипофизарно-яичниковой системы при центральном гипогонадизме у женщин. // Вопросы гинекологии, акушерства и перинатологии. – 2008. – V.7(5) – P.22–28.
9. Seminara S.B., Hayes F.J., Crowley Jr W.F. Gonadotropinreleasing hormone defi ciency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann’s syndrome): pathophysiological and genetic considerations. // Endocr. Rev. – 1998. – V.19(5). – P.521–539. DOI:10.1210/edrv.19.5.0344
10. Boehm U., Bouloux P-M., Dattani M.T., de Roux N., Dodé C., Dunkel L.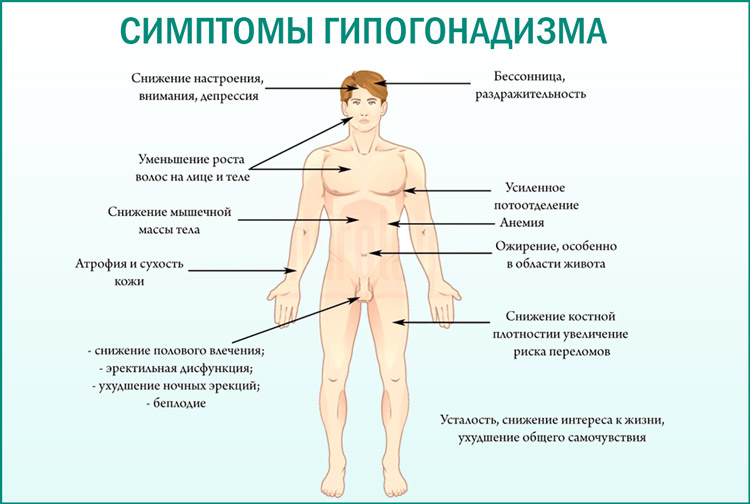 et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. // Nat. Rev. Endocrinol. – 2015. – V.11(9). – P. 547–564. DOI: 10.1038/nrendo.2015.112
et al. Expert consensus document: European Consensus Statement on congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis and treatment. // Nat. Rev. Endocrinol. – 2015. – V.11(9). – P. 547–564. DOI: 10.1038/nrendo.2015.112
11. Wray S., Grant P., Gainer H. Evidence that cells expressing luteinizing hormone-releasing hormone mRNA in the mouse are derived from progenitor cells in the olfactory placode. // Proc. Natl. Acad. Sci. – 1989. – V.86(20). – P.8132–8136. DOI:10.1073/pnas.86.20.8132
12. Shin S.-J. Sul Y., Kim J.H., Cho J.H., Kim G-H., Kim J.H., et al. Clinical, endocrinological, and molecular characterization of Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism: a single center experience. // Ann. Pediatr. Endocrinol. Metab. – 2015. – V.20(1). – P.27. DOI: 10.6065/apem.2015.20.1.27
13. Stamou M.I., Georgopoulos N.A. Kallmann syndrome: phenotype and genotype of hypogonadotropic hypogonadism. // Metabolism – 2018. – V.86. – P.124–134. DOI: 10.1016/j.metabol.2017.10.012
14. Dye A.M., Nelson G.B., Diaz-Th omas A. Delayed puberty. // Pediatr. Ann. – 2018. – V.47(1). – P.e16–e22. DOI: 10.3928/19382359-20171215-01
15. Stamou M.I., Cox K.H., Crowley Jr W.F. Discovering genes essential to the hypothalamic regulation of human reproduction using a human disease model: adjusting to life in the “-omics” era. // Endocr. Rev. – 2015. – V.2016(1). – P.4–22. DOI: 10.1210/er.2015-1045
16. Topaloğlu A.K. Update on the Genetics of Idiopathic Hypogonadotropic Hypogonadism. // J. Clin. Res. Pediatr. Endocrinol. – 2017. – V.9(Suppl 2). – P. 113–122. DOI: 10.4274/jcrpe.2017.s010
// J. Clin. Res. Pediatr. Endocrinol. – 2017. – V.9(Suppl 2). – P. 113–122. DOI: 10.4274/jcrpe.2017.s010
17. Cassatella D., Howard S. R., Acierno J.S., Xu C., Papadakis G.E., Santoni F.A. et al. Congenital hypogonadotropic hypogonadism and constitutional delay of growth and puberty have distinct genetic architectures. // Eur. J. Endocrinol. – 2018. – V.178(4). – P.377–388. DOI: 10.1530/eje-17-0568
18. Енева Н.Г., Нефедова Л.Н., Локтионова А.С., Хусниярова К.А., Иловайская И.А., Ким А.И. Проблема женского бесплодия: поиск генетических маркеров. // Журнал общей биологии – 2017. – V.78(2). – P.3–13. DOI: 10.1134/s2079086418030040
19. Franco B. Guioli S., Pragliola A., Incerti B., Bardoni B., Tonlorenzi R., et al. A gene deleted in Kallmann’s syndrome shares homology with neural cell adhesion and axonal pathfi nding molecules. // Nature – 1991. – V.353(6344). – P.529. DOI: 10.1038/353529a0
20. Sykiotis G.P., Plummer L., Hughes V.A., Au M., Durrani S., Nayak-Young S., et al. Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. // Proc. Natl. Acad. Sci. – 2010. – V.107(34). – P.15140–15144. DOI: 10.1073/pnas.1009622107
21. Hardelin J., Julliard A.K., Moniot B., Soussi‐Yanicostas N., Verney C., Schwanzel‐Fukuda M. et al. Anosmin‐1 is a regionally restricted component of basement membranes and interstitial matrices during organogenesis: implications for the developmental anomalies of X chromosome‐linked Kallmann syndrome. // Dev. Dyn. an Off . Publ. Am. Assoc. Anat. – 1999. – V.215(1). – P.26–44. DOI: 10.1002/(sici)1097-0177(199905)215:13. 3.co;2-4
3.co;2-4
22. Pitteloud N., Meysing A., Quinton R., Acierno J.S., Dwyer A.A., Plummer L. et al. Mutations in fi broblast growth factor receptor 1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. // Mol. Cell. Endocrinol. – 2006. – V.254. – P. 60–69. DOI: 10.1016/j.mce.2006.04.021
23. Falardeau J., Chung W.C.J., Beenken A., Raivio T., Plummer L., Sidis Y. et al. Decreased FGF8 signaling causes defi ciency of gonadotropin-releasing hormone in humans and mice. // J. Clin. Invest. Am Soc Clin Investig – 2008. – V.118 (8). – P. 2822–2831. DOI: 10.1172/jci34538
24. Miraoui H. Dwyer A.A., Sykiotis G.P., Plummer L., Chung W., Feng B., et al. Mutations in FGF17, IL17RD, DUSP6, SPRY4, and FLRT3 are identified in individuals with congenital hypogonadotropic hypogonadism. // Am. J. Hum. Genet. – 2013. – V. 92(5). – P. 725–743. DOI: 10.1016/j.ajhg.2013.04.008
25. Prosser H.M., Bradley A., Caldwell M.A. Olfactory bulb hypoplasia in Prokr2 null mice stems from defective neuronal progenitor migration and differentiation. // Eur. J. Neurosci. – 2007. – V. 26(12). – P. 3339–3344. DOI: 10.1111/j.1460-9568.2007.05958.x
26. Cerrato F., Shagoury J., Kralickova M., Dwyer A., Falardeau J., Ozata M. et al. Coding sequence analysis of GNRHR and GPR54 in patients with congenital and adult-onset forms of hypogonadotropic hypogonadism // Eur. J. Endocrinol. – 2006. – V. 155(suppl_1). – P. S3–S10. DOI: 10.1530/eje.1.02235
27. Chan Y.-M., de Guillebon A. , Lang-Muritano M., Plummer L., Cerrato F., Tsiaras S., et al. GNRh2 mutations in patients with idiopathic hypogonadotropic hypogonadism. // Proc. Natl. Acad. Sci. – 2009. – V.106 (28). – P. 11703–11708. DOI: 10.1073/pnas.0903449106
, Lang-Muritano M., Plummer L., Cerrato F., Tsiaras S., et al. GNRh2 mutations in patients with idiopathic hypogonadotropic hypogonadism. // Proc. Natl. Acad. Sci. – 2009. – V.106 (28). – P. 11703–11708. DOI: 10.1073/pnas.0903449106
28. Seminara S.B., Messager S., Chatzidaki E.E., Th resher R.R., Acierno J.S., Shagoury J.K., et al. Th e GPR54 gene as a regulator of puberty. // N. Engl. J. Med. – 2003. – V. 349 (17). – P. 1614–1627. DOI: 10.1056/nejmoa035322
29. de Roux N. Genin E., Carel J-C., Matsuda F., Chaussain J-L., Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. // Proc. Natl. Acad. Sci. – 2003. – V. 100(19). – P. 10972–10976. DOI: 10.1073/pnas.1834399100
30. Topaloglu A.K., Tello J.A., Kotan L.D., Ozbek M.N., Yilmaz M.B., Erdogan S. et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. // N. Engl. J. Med. – 2012. – Vol.366(7). – P.629–635. DOI: 10.1097/ogx.0b013e31825bc1be
31. Murphy K.G. Kisspeptins: regulators of metastasis and the hypothalamic‐pituitary‐gonadal axis. // J. Neuroendocrinol. – 2005. – V. 17(8). – P. 519–525. DOI: 10.1111/j.1365-2826.2005.01328.x
32. Navarro V.M., Gottsch M.L., Chavkin C., Okamura H., Clifton D.K., Steiner R.A. Regulation of gonadotropin-releasing hormone secretion by kisspeptin/dynorphin/neurokinin B neurons in the arcuate nucleus of the mouse. // J. Neurosci. – 2009. – V.29(38). – P. 11859–11866. DOI: 10.1523/jneurosci.1569-09.2009
33..gif) George J.T., Veldhuis J.D., Tena-Sempere M., Millar R.P., Anderson R.A. Exploring the pathophysiology of hypogonadism in men with type 2 diabetes: kisspeptin‐10 stimulates serum testosterone and LH secretion in men with type 2 diabetes and mild biochemical hypogonadism. // Clin. Endocrinol. – 2013. – V. 79(1). – P. 100–104. DOI: 10.1111/cen.12103
George J.T., Veldhuis J.D., Tena-Sempere M., Millar R.P., Anderson R.A. Exploring the pathophysiology of hypogonadism in men with type 2 diabetes: kisspeptin‐10 stimulates serum testosterone and LH secretion in men with type 2 diabetes and mild biochemical hypogonadism. // Clin. Endocrinol. – 2013. – V. 79(1). – P. 100–104. DOI: 10.1111/cen.12103
34. Cortés M.E., Carrera B., Rioseco H., Pablo del Río J., Vigil P. Th e role of kisspeptin in the onset of puberty and in the ovulatory mechanism: a mini-review. // J. Pediatr. Adolesc. Gynecol. – 2015. – V.28(5). – P. 286–291. DOI: 10.1016/j.jpag.2014.09.017
35. Strobel A., Issad T., Camoin L., Ozata M., Strosberg A.D. A leptin missense mutation associated with hypogonadism and morbid obesity. // Nature Genetics – 1998. – V. 18 (3). – P. 213. DOI: 10.1038/ng0398-213
36. Cravo R.M., Margatho L.O., Osborne-Lawrence S., Donato J., Atkin S., Bookout A.L. et al. Characterization of Kiss1 neurons using transgenic mouse models. // Neuroscience – 2011. – V. 173. – P. 37–56. DOI: 10.1016/j.neuroscience.2010.11.022
37. Donato Jr J., Cravo R.M., Frazão R., Elias C.F. Hypothalamic sites of leptin action linking metabolism and reproduction. // Neuroendocrinology. – 2011. – V. 93 (1). – P. 9–18. DOI: 10.1159/000322472
38. Perry J.R.B., Day F., Elks C.E., Sulem P., Thompson D.J., Ferreira T. et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. // Nature. – 2014. – V. 514(7520). – P. 92. DOI: 10.1038/nature13545
39. Perry J.R.B., Corre T., Esko T., Chasman D.I., Fischer K., Franceschini N. et al. A genome-wide association study of early menopause and the combined impact of identifi ed variants // Hum. Mol. Genet. – 2013. – V.22(7). – P. 1465–1472. DOI: 10.1093/hmg/dds551
Perry J.R.B., Corre T., Esko T., Chasman D.I., Fischer K., Franceschini N. et al. A genome-wide association study of early menopause and the combined impact of identifi ed variants // Hum. Mol. Genet. – 2013. – V.22(7). – P. 1465–1472. DOI: 10.1093/hmg/dds551
40. Caronia L.M., Martin C., Welt C.K., Sykiotis G.P., Quinton R., Th ambundit A. et al. A genetic basis for functional hypothalamic amenorrhea. // N. Engl. J. Med. – 2011. – V. 364(3). – P. 215–225. DOI: 10.1056/nejmoa0911064
41. Zhu J., Choa RE-Y., Guo M.H., Plummer L., Buck C., Palmert M.R. et al. A shared genetic basis for self-limited delayed puberty and idiopathic hypogonadotropic hypogonadism. // J. Clin. Endocrinol. Metab. – 2015. – V. 100(4). – P. E646–E654. DOI: 10.1210/jc.2015-1080
42. Dwyer A.A., Hayes F.J., Plummer L., Pitteloud N., Crowley W.F. Th e long-term clinical follow-up and natural history of men with adult-onset idiopathic hypogonadotropic hypogonadism. // J. Clin. Endocrinol. Metab. – 2010. – V. 95(9). – P. 4235–4243. DOI: 10.1016/s0022-5347(11)60183-3
43. Gordon C.M., Ackerman K.E., Berga S.L., Kaplan J.R., Mastorakos G., Misra M. et al. Functional hypothalamic amenorrhea: an endocrine society clinical practice guideline. // J. Clin. Endocrinol. Metab. – 2017. – V. 102(5). – P. 1413–1439. DOI: 10.1210/jc.2017-00131
44. Gordon C.M. Functional hypothalamic amenorrhea. // N. Engl. J. Med. – 2010. – V. 363(4). – P. 365–371. DOI: 10.1056/nejmcp0912024
45. Sidhoum V.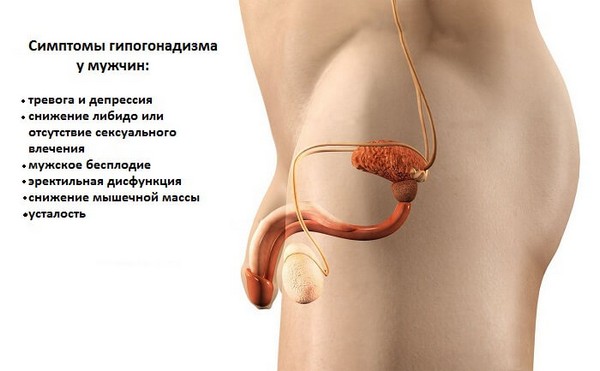 F., Chan Y-M., Lippincott M.F., Balasubramanian R., Quinton R., Plummer L. et al. Reversal and relapse of hypogonadotropic hypogonadism: Resilience and fragility of the reproductive neuroendocrine system. // J. Clin. Endocrinol. Metab. – 2014. – V. 99(3). – P. 861–870. DOI: 10.1210/jc.2013-2809
F., Chan Y-M., Lippincott M.F., Balasubramanian R., Quinton R., Plummer L. et al. Reversal and relapse of hypogonadotropic hypogonadism: Resilience and fragility of the reproductive neuroendocrine system. // J. Clin. Endocrinol. Metab. – 2014. – V. 99(3). – P. 861–870. DOI: 10.1210/jc.2013-2809
46. Dwyer A.A., Raivio T., Pitteloud N. Management of endocrine disease: reversible hypogonadotropic hypogonadism. // Eur. J. Endocrinol. – 2016. – V. 174(6). – P. R267–R274. DOI: 10.1530/eje-15-1033
47. Meczekalski B., Genazzani A.R., Genazzani A.D., WarenikSzymankiewicz A., Luisi M. Clinical evaluation of patients with weight loss-related amenorrhea: neuropeptide Y and luteinizing hormone pulsatility. // Gynecol. Endocrinol. – 2006. – V. 22(5). – P. 239–243. DOI: 10.1080/14767050600761992
48. Tolle V., Kadem M., Bluet-Pajot M-T., Frere D., Foulon C., Bossu C., et al. Balance in ghrelin and leptin plasma levels in anorexia nervosa patients and constitutionally thin women. // J. Clin. Endocrinol. Metab. – 2003. – V. – 88(1). – P. – 109–116. DOI: 10.1210/jc.2002-020645
49. Rivier C., Brownstein M., Spiess J., Rivier J., Vale W. In vivo corticotropin-releasing factor-induced secretion of adrenocorticotropin, β-endorphin, and corticosterone. // Endocrinology. – 1982. – V. 110(1). – P. 272–278. DOI: 10.1210/endo-110-1-272
50. Remorgida V., Venturini P.L., Anserini P., Salerno E., De Cecco L. Naltrexone in functional hypothalamic amenorrhea and in the normal luteal phase. // Obstet. Gynecol. – 1990. – V. 76(6). – P. 1115–1120. Доступно по ссылке: https://www. ncbi.nlm.nih.gov/pubmed/2122343. Ссылка активна на 4.11.2019
ncbi.nlm.nih.gov/pubmed/2122343. Ссылка активна на 4.11.2019
51. Genazzani A.D., Luisi M., Malavasi B., Strucchi C., Luisi S., Casarosa E. et al. Pulsatile secretory characteristics of allopregnanolone, a neuroactive steroid, during the menstrual cycle and in amenorrheic subjects. // Eur. J. Endocrinol. – 2002. – V. 146(3). – P. 347–356. DOI: 10.1530/eje.0.1460347
52. Michopoulos V., Mancini F., Loucks T.L., Berga S.L. Neuroendocrine recovery initiated by cognitive behavioral therapy in women with functional hypothalamic amenorrhea: a randomized, controlled trial. // Fertil. Steril. – 2013. – V. 99(7). – P. 2084–2091. DOI: 10.3410/f.717995501.793475957
53. Иловайская И.А. Опухолевые и неопухолевые заболевания гипофиза и репродуктивная система. // Acta Biomed. Sci. – 2012. – №3 (85), часть 1. Доступно по ссылке: https://www.actabiomedica.ru/jour/article/view/871. Ссылка активна на 4.11.2019
54. Warren M.P., Vu C. Central causes of hypogonadismfunctional and organic. // Endocrinol. Metab. Clin. North Am. – 2003. – V.32(3). – P. 593–612. DOI: 10.1016/s0889-8529(03)00042-2
55. Unuane D., Tournaye H., Velkeniers B., Poppe K. Endocrine disorders & female infertility. // Best Pract. Res. Clin. Endocrinol. Metab. – 2011. – V. 25(6). – P. 861–873. DOI: 10.1016/j.beem.2011.08.001
56. Bergh T., Skarin G., Nillius S.J., Wide L. Pulsatile GnRH therapy–an alternative successful therapy for induction of ovulation in infertile normo-and hyperprolactinaemic amenorrhoeic women with pituitary tumours. // Eur. J. Endocrinol. – 1985. – V. 110(4). – P. 440–444. DOI: 10.1530/acta.0.1100440
// Eur. J. Endocrinol. – 1985. – V. 110(4). – P. 440–444. DOI: 10.1530/acta.0.1100440
57. Kaltsas G.A., Mukherjee J.J., Jenkins P.J., Satta M.A., Islam N., Monson J.P. et al. Menstrual irregularity in women with acromegaly. // J. Clin. Endocrinol. Metab. – 1999. – V. 84(8). – P. 2731–2735. DOI: 10.1210/jcem.84.8.5858
58. Newell-Price J., Grossman A.B. Diff erential diagnosis of Cushing’s syndrome. // Arq. Bras. Endocrinol. Metabol. – 2007. – V. 51(8). – P. 1199–1206. DOI: 10.1590/s0004- 27302007000800005
59. Ferreira L., Silveira G., Latronico A.C. Approach to the patient with hypogonadotropic hypogonadism. // J. Clin. Endocrinol. Metab. – 2013. – V. 98(5). – P. 1781–1788. DOI: 10.1210/jc.2012-3550
60. Faje A. Hypophysitis: evaluation and management. // Clin. diabetes Endocrinol. – 2016. – V. 2(1). – P. 15. DOI: 10.1186/s40842-016-0034-8
Гипогонадизм у женщин | DermNet NZ
Автор: д-р Гэвин Эссон, стажер Фонда, NHS Lothian, Эдинбург, Шотландия. Главный редактор DermNet NZ: адъюнкт. Доц. Проф. Аманда Окли, дерматолог, Гамильтон, Новая Зеландия. Апрель 2019.
Что такое гипогонадизм у женщин?
Гипогонадизм у женщин — это нарушение функции яичников с нарушением производства половых клеток (яиц) и половых гормонов (эстроген и прогестерон).
- Первичный гипогонадизм относится к состоянию яичников (первичная недостаточность яичников / гипергонадотропный гипогонадизм).
- Вторичный гипогонадизм относится к отказу гипоталамуса или гипофиза (гипогонадотропный гипогонадизм).


Что вызывает гипогонадизм у женщин?
Гипогонадизм у женщин возникает из-за нарушения любого участка пути оси гипоталамус – гипофиз – яичники (рис. 1). В правильно функционирующем пути оси гипоталамус-гипофиз-яичники:
- Гипоталамус вырабатывает гонадотропин-высвобождающий гормон (ГнРГ) в начале полового созревания
- Затем гонадолиберин действует на гипофиз, который производит фолликулостимулирующий гормон (ФСГ) и лютеинизирующий гормон (ЛГ).
- ФСГ и ЛГ воздействуют на яичники, стимулируя выработку эстрогена и прогестерона.
Затем
Рисунок 1. Осевой путь гипоталамус-гипофиз-яичники
ФСГ, стимулированный фолликулами; ГнРГ, гонадотропин-рилизинг-гормон: ЛГ, лютеинизирующий гормон.
Кто болеет гипогонадизмом?
Первичная недостаточность яичников и вторичный гипогонадизм могут быть врожденными или приобретенными [1,2].
Врожденная первичная недостаточность яичников
Основной механизм врожденной первичной недостаточности яичников в большинстве случаев остается неизвестным.Некоторые случаи касаются:
Приобретенной первичной недостаточности яичников
Причины приобретенной первичной недостаточности яичников включают:
- Лекарства, такие как хлорамбуцил, циклофосфамид и алкилирующие агенты
- Радиотерапия
- Аутоиммунные болезни, включая аутоиммунный полигландулярный синдром 1 типа
- Вирусные инфекции, включая оофорит паротита, туберкулез (ТБ), малярию, ветряную оспу и
- Бактериальные инфекции, такие как Shigella
- Ятрогенное заболевание, например проблемы после овариэктомии (хирургическое удаление яичников).
Врожденный вторичный гипогонадизм
Врожденный вторичный гипогонадизм — это недостаточность гонадотропинов, вызванная генетической мутацией, например, при синдроме Каллмана.
Приобретенный вторичный гипогонадизм
Приобретенный вторичный гипогонадизм может быть следствием поражения гипофиза / гипоталамуса. Причины приобретенного вторичного гипогонадизма могут включать:
- Внутричерепные объемные образования (например, опухоли и кисты)
- Инфильтративная болезнь (например, саркоидоз и гемохроматоз)
- Инфекция (например, менингит и туберкулез)
- Апоплексия гипофиза (кровотечение в гипофиз)
- Травма.
Гонадотропины могут подавляться:
- Хроническими заболеваниями (например, диабетом, анорексией, ожирением и заболеванием почек)
- Чрезмерные упражнения
- Критическая болезнь
- Хроническое употребление опиатов, глюкокортикоидов или анаболических стероидов
- Гиперпролактинемия (избыток гормона пролактина, индуцирующего молоко).
Каковы клинические признаки гипогонадизма у женщин?
Клинические признаки гипогонадизма зависят от возраста обращения [3].
Недостаточность эстрогена до полового созревания
Симптомы низкого уровня эстрогена редко присутствуют при гипогонадизме до полового созревания. Признаки: отсутствие полового созревания (замедленный рост и отсутствие волос на лобке) и первичная аменорея (отсутствие менархе).
Дефицит эстрогена после завершения полового созревания
После завершения полового созревания признаки гипогонадизма включают:
- Вторичная аменорея (прекращение регулярных менструаций на 3 месяца или прекращение нерегулярных менструаций на 6 месяцев)
- Симптомы климактерического периода (перименопауза): сердцебиение, непереносимость тепла, приливы, ночная потливость, раздражительность, беспокойство, депрессия, нарушение сна, потеря либидо, грубые волосы, сухость влагалища и усталость
- Бесплодие.
Каковы осложнения дефицита эстрогена?
Долгосрочные риски дефицита эстрогена включают повышенный риск остеопороза и сердечно-сосудистых заболеваний. Риск возрастает с возрастом начала заболевания. Напротив, риск рака груди может быть немного снижен.
Какие изменения кожи могут быть вызваны гипогонадизмом у женщин?
Эстроген играет ключевую роль в поддержании здоровья кожи. Эстроген помогает поддерживать толщину кожи и уровень коллагена, эластичность и влажность кожи.Также считается, что он играет роль в заживлении ран [4].
Низкий уровень эстрогенов связан с:
Кожные изменения также могут отражать первопричину гипогонадизма; например, гиперпигментация может быть признаком аутоиммунного заболевания.
Как диагностировать гипогонадизм?
При подозрении на гипогонадизм после подробного анамнеза и обследования можно выбрать следующий путь исследования.
- Пациента направить к специалисту.
- Первоначальные исследования должны включать [5,6]:
- Хорионический гонадотропин человека (ХГЧ) исключает беременность
- FSH и LH
- Эстрадиол
- Тиреотропный гормон (ТТГ) — заболевания щитовидной железы могут проявляться аменореей
- Пролактин сыворотки
- Ультразвуковое исследование органов малого таза — до полового созревания.
Интерпретация результатов
- Положительный результат теста на ХГЧ обусловлен беременностью.
- Если матка отсутствует или имеются другие анатомические аномалии, необходимо провести исследование кариотипа.
- Низкие или низкие / нормальные уровни ФСГ и ЛГ могут указывать на гипогонадотропный гипогонадизм. Следует рассмотреть возможность проведения МРТ головного мозга.
- До полового созревания, высокие уровни ФСГ и ЛГ могут указывать на первичную недостаточность яичников. Следует оценить гормоны надпочечников и провести анализ кариотипа.
- Повышенный результат пролактина должен побуждать к рассмотрению МРТ гипофиза.
Как лечить гипогонадизм у женщин?
Лечение гипогонадизма направлено, по возможности, на лежащую в основе патологию, помогая женщине стать фертильной при желании и предотвращая долгосрочные осложнения гипоэстрогении (например, остеопороз, сердечно-сосудистые заболевания и урогенитальную атрофию).
Как правило, женщины репродуктивного возраста с гипоэстрогенизмом должны получать заместительную гормональную терапию. Следует запросить мнение специалиста, так как при гормональной терапии могут возникнуть серьезные осложнения, такие как:
- Гиперплазия эндометрия и карцинома при безальтернативной терапии эстрогенами — обычно добавляют прогестин, чтобы остановить это
- Повышенный риск рака груди, венозной тромбоэмболии, инсульта и ишемической болезни сердца.
После менопаузы заместительная гормональная терапия показана при значительных симптомах.
Гипогонадизм — AMBOSS
Последнее обновление: 26 октября 2020 г.
Резюме
Гипогонадизм — это клинический синдром, связанный с нарушением функциональной активности гонад. Могут быть затронуты как мужчины, так и женщины. Он классифицируется как первичный или вторичный: первичный гипогонадизм (гипергонадотропный гипогонадизм) обычно вызывается врожденными нарушениями полового развития, поражающими гонады (например, синдром Тернера, синдром Клайнфельтера) или приобретенным повреждением гонад (например, синдром Тернера, синдром Клайнфельтера).г., облучение, инфицирование). Вторичный гипогонадизм (гипогонадотропный гипогонадизм) чаще всего вызывается нарушениями гипофиза или гипоталамуса (например, краниофарингиомой, синдромом Каллмана). Характерные признаки у мужчин включают гипоплазию яичек, гинекомастию и отсутствие роста волос на лице, в то время как у женщин обычно наблюдается аменорея. После клинической оценки диагноз подтверждается тестами на гормоны, и может быть рассмотрен вопрос о генетическом тестировании. Лечение включает устранение основной причины и заместительную гормональную терапию.
Этиология
Гипогонадизм — это клинический синдром, связанный с нарушением функциональной активности гонад.
Гипергонадотропный гипогонадизм вызывается недостаточной выработкой половых стероидов в половых железах.
Гипогонадотропный гипогонадизм вызывается недостаточным высвобождением гонадотропин-рилизинг-гормона (ГнРГ) и / или высвобождением гонадотропина в гипоталамо-гипофизарной оси.
Патофизиология
Снижение функциональной активности гонад → снижение биосинтеза половых гормонов → нарушение вторичных половых признаков и бесплодие
Клинические признаки
Диагностика
- Текущие испытания
- Дальнейшие исследования: на основании подозреваемой этиологии
Лечение
Ссылки
Байларджон Дж., Раджи М.А., Урбан Р.Дж. и др.Гипогонадизм, индуцированный опиоидами, в Соединенных Штатах. Протоколы клиники Мэйо. Инновации, качество и результаты . 2019; 3
(3): с.276-284.
DOI: 10.1016 / j.mayocpiqo.2019.06.007. | Открыть в режиме чтения QxMD
Гипогонадотропный гипогонадизм — обзор
Синдром Каллмана
HH, связанный с ненормальным обонянием (аносмия / гипосмия), называется синдромом Каллмана. Как и в случае ГГ и нормального обоняния, у людей с СК могут наблюдаться другие нерепродуктивные клинические признаки, включая ихтиоз, атрезию хоан, подковообразные почки и зеркальные движения рук (синкинезия). 67 Несколько генов, критических для функции оси HPG и развития обоняния, были идентифицированы посредством исследования синдрома Каллмана. В частности, мутации синдрома Каллмана 1 ( KAL-1) 82 , 83 и рецептора фактора роста фибробластов 1 ( FGFR1) 84 вовлечены в Х-сцепленные и аутосомно-доминантные формы. болезни, соответственно, и, по-видимому, составляют примерно 20% пациентов с СК. 85 Мутации в гене рецептора прокинетицина-2 ( PROKR2) , рецептора, связанного с G-белком, и в его лиганде прокинетицин-2 (PROK2) также были идентифицированы у пациентов с KS, 85 , демонстрируя, что Передача сигналов prokineticin важна для развития обонятельной системы и оси HPG. Один из пациентов в этой начальной серии был гетерозиготным по мутации PROKR2 и KAL1 , что указывает на возможный дигенный тип наследования. 85 Наконец, мутации в факторе рилизинг-гормона носового эмбриона лютеинизирующего гормона ( NELF ), который играет роль в миграции нейронов GnRH и разрастании обонятельных аксонов, 86 также вовлечены в патогенез KS. 87 Гетерозиготная делеция в NELF первоначально была описана как компонент, наряду с FGFR1, дигенного KS, 88 , но недавно сообщалось, что NELF может приводить к HH или KS через моногенные, а также дигенные наследование. 89
Различия между различными аномалиями пубертатного развития не всегда четкие. Например, комплексное исследование PROK2 и PROKR2 у пациентов с HH и KS обнаружило мутации в обоих генах, распределенных в обеих группах пациентов. 90 Мутации в гене FGF8 , который кодирует лиганд для FGFR1, наблюдались у пациентов с HH, сопровождаемых различными обонятельными фенотипами. 91 Мутации в CHD7 , гене, ответственном за синдром CHARGE, который имеет некоторые общие черты развития с KS, были идентифицированы у пациентов как с нормосмическим HH, так и с KS. 92 , 93 Совсем недавно мутации в FGFR1 , FGF8 и PROKR2 были выявлены среди субъектов с комбинированным дефицитом гормона гипофиза (CPHD) и септооптической дисплазией (SOD), что указывает на то, что в некоторых случаях HH и CPHD / SOD могут иметь общую генетическую этиологию. 94 , 95 Различие между HH и конституциональной задержкой роста и полового созревания (CDGP) также не всегда ясно. Вариация FGFR1 , по-видимому, не является основной причиной CDGP, 81 , хотя мутации с потерей функции в FGFR1 могут вызывать задержку полового созревания у представителей родословных HH. 96 , 97 Сообщалось также о случаях обратимого HH, 98 повышая вероятность того, что некоторые случаи HH могут представлять тяжелые версии CDGP, еще больше стирая различие между HH и CDGP.
Европейское консенсусное заявление о врожденном гипогонадотропном гипогонадизме — патогенез, диагностика и лечение
Бьянко, С. Д. и Кайзер, У. Б. Генетические и молекулярные основы идиопатического гипогонадотропного гипогонадизма. Nat. Rev. Endocrinol. 5 , 569–576 (2009).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Schwanzel-Fukuda, M., Bick, D. & Pfaff, D. W. Клетки, экспрессирующие лютеинизирующий гормон-рилизинг-гормон (LHRH), не мигрируют нормально при наследственном гипогонадальном (Kallmann) синдроме. Brain Res. Мол. Brain Res. 6 , 311–326 (1989).
Артикул
CAS
PubMed
Google Scholar
Тейшейра, Л. и др. . Дефектная миграция нейроэндокринных клеток GnRH в арринэнцефалических состояниях человека. J. Clin. Вкладывать деньги. 120 , 3668–3672 (2010).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Семинара, С. Б., Хейс, Ф. Дж. И Кроули, В. Ф. Младший. Дефицит гонадотропин-рилизинг-гормона у человека (идиопатический гипогонадотропный гипогонадизм и синдром Каллмана): патофизиологические и генетические соображения. Endocr. Ред. 19 , 521–539 (1998).
CAS
PubMed
Google Scholar
Митчелл, А. Л., Дуайер, А., Питтелуд, Н. и Куинтон, Р. Генетические основы и вариабельное фенотипическое выражение синдрома Каллмана: к объединяющей теории. Trends Endocrinol. Метаб. 22 , 249–258 (2011).
CAS
PubMed
Google Scholar
Райвио Т. и др. . Лечение идиопатического гипогонадотропного гипогонадизма. N. Engl. J. Med. 357 , 863–873 (2007).
Артикул
CAS
PubMed
Google Scholar
Сидхум, В. Ф. и др. . Обратный ход и рецидив гипогонадотропного гипогонадизма: устойчивость и хрупкость репродуктивной нейроэндокринной системы. J. Clin. Эндокринол. Метаб. 99 , 861–870 (2014).
Артикул
CAS
PubMed
Google Scholar
Рей, Р. А. и др. . Мужской гипогонадизм: расширенная классификация, основанная на подходе, основанном на эндокринной физиологии развития. Андрология 1 , 3–16 (2013).
Артикул
CAS
PubMed
Google Scholar
Чан, Ю. М. Влияние кисспептина на секрецию гормонов у людей. Adv. Exp. Med. Биол. 784 , 89–112 (2013).
Артикул
CAS
PubMed
Google Scholar
Дуайер, А. А. и др. . Испытание предварительной обработки рекомбинантным фолликулостимулирующим гормоном для лечения ГнРГ-индуцированной фертильности у пациентов с врожденным гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 98 , E1790 – E1795 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Сикиотис, Г.P. и др. . Олигогенная основа изолированного дефицита гонадотропин-рилизинг-гормона. Proc. Natl Acad. Sci. США 107 , 15140–15144 (2010).
Артикул
PubMed
Google Scholar
Петак С. М. и др. . Американская ассоциация клинических эндокринологов: медицинские рекомендации по клинической практике для оценки и лечения гипогонадизма у взрослых пациентов мужского пола — обновление 2002 г. Endocr. Практик. 8 , 440–456 (2002).
PubMed
Google Scholar
Ван, К. и др. . Исследование, лечение и мониторинг гипогонадизма с поздним началом у мужчин: рекомендации ISA, ISSAM, EAU, EAA и ASA. евро. J. Endocrinol. 159 , 507–514 (2008).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Бхасин, С. и др. . Терапия тестостероном у мужчин с синдромами андрогенной недостаточности: руководство по клинической практике эндокринного общества. J. Clin. Эндокринол. Метаб. 95 , 2536–2559 (2010).
Артикул
CAS
PubMed
Google Scholar
Форни, П.Э., Тейлор-Бердс, К., Мелвин, В.С., Уильямс, Т. и Рэй, С. Нервный гребень и эктодермальные клетки смешиваются в носовой плакоде, давая начало нейронам GnRH-1, сенсорным нейронам , и обонятельные обволакивающие клетки. J. Neurosci. 31 , 6915–6927 (2011).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Форни, П. Э. и Рэй, С. ГнРГ, аносмия и гипогонадотропный гипогонадизм — где мы? Фронт. Нейроэндокринол. 36 , 167–177 (2014).
Google Scholar
Dode, C. и др. .Мутации потери функции в FGFR1 вызывают аутосомно-доминантный синдром Каллмана. Nat. Genet. 33 , 463–465 (2003).
Артикул
CAS
PubMed
Google Scholar
Питтелуд, Н. и др. . Мутации рецептора 1 фактора роста фибробластов вызывают как синдром Каллмана, так и нормосмический идиопатический гипогонадотропный гипогонадизм. Proc. Natl Acad. Sci. США 103 , 6281–6286 (2006).
Артикул
CAS
PubMed
Google Scholar
Фалардо, Дж. и др. . Снижение передачи сигналов FGF8 вызывает дефицит гонадотропин-рилизинг-гормона у людей и мышей. J. Clin. Вкладывать деньги. 118 , 2822–2831 (2008).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Торнберг, Дж. и др. .Гепарансульфат-6-O-сульфотрансфераза 1, ген, участвующий во внеклеточных модификациях сахара, мутирован у пациентов с идиопатическим гипогонадотропным гипогонадизмом. Proc. Natl Acad. Sci. США 108 , 11524–11529 (2011).
Артикул
PubMed
Google Scholar
Мирауи, Х. и др. . Мутации в FGF17 , IL17RD , DUSP6 , SPRY4 и FLRT3 идентифицированы у лиц с врожденным гипогонадотропным гипогонадизмом. Am. J. Hum. Genet. 92 , 725–743 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Джилл, Дж. К., Моэнтер, С. М. и Цай, П. С. Регуляция развития нейронов гонадотропин-рилизинг-гормона с помощью передачи сигналов фактора роста фибробластов. Эндокринология 145 , 3830–3839 (2004).
Артикул
CAS
PubMed
Google Scholar
Огата, Т. и др. . Фенотип синдрома Каллмана у пациентки с синдромом CHARGE и мутацией CHD7 . Endocr. J. 53 , 741–743 (2006).
Артикул
CAS
PubMed
Google Scholar
Jongmans, M.C. и др. . CHD7 мутаций у пациентов с изначально диагностированным синдромом Каллмана — клиническое совпадение с синдромом CHARGE. Clin. Genet. 75 , 65–71 (2009).
Артикул
CAS
PubMed
Google Scholar
Ким, Х. Г. и др. . Мутации в CHD7 , кодирующем белок, ремоделирующий хроматин, вызывают идиопатический гипогонадотропный гипогонадизм и синдром Каллмана. Am. J. Hum. Genet. 83 , 511–519 (2008).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Маркос, С. и др. . Преобладание CHD7 missense по сравнению с усекающими мутациями выше у пациентов с синдромом Каллмана, чем у типичных пациентов с CHARGE. J. Clin. Эндокринол. Метаб. 99 , E2138 – E2143 (2014).
Артикул
CAS
PubMed
Google Scholar
Balasubramanian, R. et al . Функционально скомпрометированные аллели CHD7 у пациентов с изолированным дефицитом гонадолиберина. Proc. Natl Acad. Sci. США 111 , 17953–17958 (2014).
Артикул
CAS
PubMed
Google Scholar
Пинго, В. и др. . Мутации с потерей функции в SOX10 вызывают синдром Каллмана с глухотой. Am. J. Hum. Genet. 92 , 707–724 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Вааралахти, К. и др. . De novo SOX10 нонсенс-мутация у пациента с синдромом Каллмана и потерей слуха. Pediatr. Res. 76 , 115–116 (2014).
Артикул
CAS
PubMed
Google Scholar
Dode, C. и др. . Синдром Каллмана: мутации в генах, кодирующих прокинетицин-2 и рецептор прокинетицина-2. PLoS Genet. 2 , e175 (2006).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Лерой, К. и др. . Двуаллельные мутации в гене прокинетицина-2 в двух спорадических случаях синдрома Каллмана. евро. J. Hum. Genet. 16 , 865–868 (2008).
Артикул
CAS
PubMed
Google Scholar
Коул, Л. В. и др. . Мутации в генах прокинетицина 2 и рецептора прокинетицина 2 при дефиците гонадотропин-рилизинг-гормона человека: молекулярная генетика и клинический спектр. J. Clin. Эндокринол. Метаб. 93 , 3551–3559 (2008).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Абреу А. П. и др. . Мутации потери функции в генах, кодирующих прокинетицин-2 или рецептор прокинетицина-2, вызывают аутосомно-рецессивный синдром Каллмана. J. Clin. Эндокринол. Метаб. 93 , 4113–4118 (2008).
Артикул
CAS
PubMed
Google Scholar
Ким, Х.Г. и др. . WDR11, белок WD, который взаимодействует с фактором транскрипции EMX1, мутирован при идиопатическом гипогонадотропном гипогонадизме и синдроме Каллмана. Am. J. Hum. Genet. 87 , 465–479 (2010).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Куэйнор, С. Д. и др. . Распространенность дигенных мутаций у пациентов с нормосмическим гипогонадотропным гипогонадизмом и синдромом Каллмана. Fertil. Стерил. 96 , 1424–1430 (2011).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Ханчате, Н. К. и др. . SEMA3A , ген, участвующий в поиске аксонального пути, мутирован у пациентов с синдромом Каллмана. PLoS Genet. 8 , e1002896 (2012).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Янг, Дж. и др. . SEMA3A Делеция в семье с синдромом Каллмана подтверждает роль семафорина 3A в половом созревании человека и развитии обонятельной системы. Hum. Репродукция. 27 , 1460–1465 (2012).
Артикул
CAS
PubMed
Google Scholar
Кансакоски, Дж. и др. . Скрининг мутаций SEMA3A и SEMA7 A у пациентов с врожденным гипогонадотропным гипогонадизмом. Pediatr. Res. 75 , 641–644 (2014).
Артикул
CAS
PubMed
Google Scholar
Котан Л. Д. и др. . Мутации в FEZF1 вызывают синдром Каллмана. Am. J. Hum. Genet. 95 , 326–331 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Топалоглу, А.К. и др. . Инактивация мутации KISS1 и гипогонадотропный гипогонадизм. N. Engl. J. Med. 366 , 629–635 (2012).
Артикул
CAS
PubMed
Google Scholar
де Ру, Н. и др. . Гипогонадотропный гипогонадизм из-за потери функции производного KiSS1 пептидного рецептора GPR54. Proc. Natl Acad. Sci. США 100 , 10972–10976 (2003).
Артикул
CAS
PubMed
Google Scholar
Семинара, С. Б. и др. . Ген GPR54 как регулятор полового созревания. N. Engl. J. Med. 349 , 1614–1627 (2003).
Артикул
CAS
PubMed
Google Scholar
Пинилла, Л., Агилар, Э., Дьегес, К., Миллар, Р. П. и Тена-Семпере, М. Кисспептины и размножение: физиологические роли и регуляторные механизмы. Physiol. Ред. 92 , 1235–1316 (2012).
Артикул
CAS
PubMed
Google Scholar
Кумар Д. и др. . Нейроны кисспептина дугообразного ядра мыши сообщаются с нейронами гонадолиберин внутриутробно . J. Neurosci. 34 , 3756–3766 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Кларксон, Дж.& Herbison, A.E. Эстроген, кисспептин, GPR54 и преовуляторный выброс лютеинизирующего гормона. J. Neuroendocrinol. 21 , 305–311 (2009).
Артикул
CAS
PubMed
Google Scholar
Кауфман, А.С. и др. . Половая дифференциация экспрессии гена Kiss1 в головном мозге крысы. Эндокринология 148 , 1774–1783 (2007).
Артикул
CAS
PubMed
Google Scholar
Майер, К. и др. . Время и завершение полового созревания у самок мышей зависят от передачи сигналов рецептора эстрогена в нейронах кисспептина. Proc. Natl Acad. Sci. США 107 , 22693–22698 (2010).
Артикул
PubMed
Google Scholar
Топалоглу, А. К. и др. . Мутации TAC3 и TACR3 при семейном гипогонадотропном гипогонадизме раскрывают ключевую роль нейрокинина B в центральном контроле репродукции. Nat. Genet. 41 , 354–358 (2009).
Артикул
CAS
PubMed
Google Scholar
Янг, Дж. и др. . Дефекты TAC3 и TACR3 вызывают врожденный гипогонадотропный гипогонадизм гипоталамуса у людей. J. Clin. Эндокринол. Метаб. 95 , 2287–2295 (2010).
Артикул
CAS
PubMed
Google Scholar
Джанетти, Э. и др. . Мутации TAC3 / TACR3 демонстрируют преимущественную активацию высвобождения гонадотропин-рилизинг гормона нейрокинином B в неонатальном периоде жизни с последующим обращением во взрослом возрасте. J. Clin. Эндокринол. Метаб. 95 , 2857–2867 (2010).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Наварро, В. М. и Тена-Семпере, М. Нейроэндокринный контроль с помощью кисспептинов: роль в метаболической регуляции фертильности. Nat. Rev. Endocrinol. 8 , 40–53 (2012).
Артикул
CAS
Google Scholar
Грабовский, Э. и др. . Глутаматергическая и ГАМКергическая иннервация нейронов гонадотропин-рилизинг-гормона-I человека. Эндокринология 153 , 2766–2776 (2012).
Артикул
CAS
PubMed
Google Scholar
Овергаард, А., Ruiz-Pino, F., Castellano, J. M., Tena-Sempere, M. & Mikkelsen, J. D. Несопоставимые изменения в экспрессии кисспептина и нейрокинина B в дугообразном ядре после манипуляции с половыми стероидами показывают дифференциальную регуляцию двух пептидов KNDy у крыс. Эндокринология 155 , 3945–3955 (2014).
Артикул
CAS
PubMed
Google Scholar
Марголин Д. Х. и др. . Атаксия, деменция и гипогонадотропизм, вызванные нарушением убиквитинирования. N. Engl. J. Med. 368 , 1992–2003 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Синофзик, М. и др. . PNPLA6 мутации вызывают синдромы Буше-Нойхаузера и Гордона Холмса как часть широкого нейродегенеративного спектра. Мозг 137 , 69–77 (2014).
Артикул
PubMed
Google Scholar
Топалоглу, А.К. и др. . Мутации потери функции в PNPLA6 , кодирующей целевую эстеразу невропатии, лежат в основе пубертатной недостаточности и неврологических нарушений при синдроме Гордона Холмса. J. Clin. Эндокринол. Метаб. 99 , E2067 – E2075 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Тата Б. и др. . Гаплонедостаточность Dmxl2 , кодирующего синаптический белок, вызывает бесплодие, связанное с потерей нейронов GnRH у мышей. PLoS Biol. 12 , e1001952 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Фаруки, И. С. и О’Рахилли, С. Мутации лигандов и рецепторов пути лептин-меланокортин, которые приводят к ожирению. Nat. Clin. Практик Эндокринол. Метаб. 4 , 569–577 (2008).
Артикул
CAS
PubMed
Google Scholar
Квеннелл, Дж.Х. и др. . Лептин косвенно регулирует функцию нейронов гонадотропин-рилизинг-гормона. Эндокринология 150 , 2805–2812 (2009).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Донато Дж. Младший и др. . Эффект лептина на половое созревание у мышей передается вентральным премамиллярным ядром и не требует передачи сигналов в нейронах Kiss1. J. Clin.Вкладывать деньги. 121 , 355–368 (2011).
Артикул
PubMed
Google Scholar
Bellefontaine, N. et al. . Лептин-зависимая нейрональная передача сигналов NO в преоптическом гипоталамусе способствует размножению. J. Clin. Вкладывать деньги. 124 , 2550–2559 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Куири-Ханнинен, Т. и др. . Повышенная активность оси гипоталамус-гипофиз-яички в младенчестве приводит к усилению действия андрогенов у недоношенных мальчиков. J. Clin. Эндокринол. Метаб. 96 , 98–105 (2011).
Артикул
CAS
PubMed
Google Scholar
Куири-Ханнинен, Т. и др. . Постнатальные изменения в оси гипофиз – яичник у недоношенных и доношенных девочек. Дж.Clin. Эндокринол. Метаб. 96 , 3432–3439 (2011).
Артикул
CAS
PubMed
Google Scholar
Сиск, К. Л. и Фостер, Д. Л. Нейронные основы полового созревания и подросткового возраста. Nat. Neurosci. 7 , 1040–1047 (2004).
Артикул
CAS
PubMed
Google Scholar
Бояр Р. М. и др. .Половое созревание человека. Одновременное усиление секреции лютеинизирующего гормона и тестостерона во время сна. J. Clin. Вкладывать деньги. 54 , 609–618 (1974).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Wu, F. C., Butler, G. E., Kelnar, C. J. и Sellar, R. E. Особенности пульсирующей секреции лютеинизирующего гормона до и во время наступления половой зрелости у мальчиков: исследование с использованием иммунорадиометрического анализа. J. Clin. Эндокринол. Метаб. 70 , 629–637 (1990).
Артикул
CAS
PubMed
Google Scholar
Дункель Л. и др. . Изменения в развитии 24-часовых профилей лютеинизирующего гормона и фолликулостимулирующего гормона от препубертата до середины полового созревания у мальчиков. J. Clin. Эндокринол. Метаб. 74 , 890–897 (1992).
Артикул
CAS
PubMed
Google Scholar
Валерий, К., Schteingart, H. F. & Rey, R. A. Препубертатное яичко: биомаркеры и функции. Curr. Opin. Эндокринол. Диабет Ожирение. 20 , 224–233 (2013).
Артикул
CAS
PubMed
Google Scholar
Сехестед, А. и др. . Сывороточные ингибин А и ингибин В у здоровых девочек препубертатного, пубертатного и подросткового возраста и взрослых женщин: зависимость от возраста, стадии полового созревания, менструального цикла, уровней фолликулостимулирующего гормона, лютеинизирующего гормона и эстрадиола. J. Clin. Эндокринол. Метаб. 85 , 1634–1640 (2000).
CAS
PubMed
Google Scholar
Андерссон, А. М. и др. . Сывороточный ингибин B у здоровых мальчиков пубертатного и подросткового возраста: зависимость от возраста, стадии полового созревания и уровней фолликулостимулирующего гормона, лютеинизирующего гормона, тестостерона и эстрадиола. J. Clin. Эндокринол. Метаб. 82 , 3976–3981 (1997).
CAS
PubMed
Google Scholar
Хаген, К.P. и др. . Уровни антимюллерова гормона в сыворотке как маркер функции яичников у 926 здоровых женщин от рождения до зрелого возраста и у 172 пациентов с синдромом Тернера. J. Clin. Эндокринол. Метаб. 95 , 5003–5010 (2010).
Артикул
CAS
PubMed
Google Scholar
Аксглаеде Л. и др. . Изменения антимюллеровского гормона (АМГ) на протяжении жизни: популяционное исследование 1027 здоровых мужчин от рождения (пуповинная кровь) до возраста 69 лет. J. Clin. Эндокринол. Метаб. 95 , 5357–5364 (2010).
Артикул
CAS
PubMed
Google Scholar
Ивелл Р., Хенг К. и Ананд-Ивелл Р. Инсулиноподобный фактор 3 и ось HPG у мужчин. Фронт. Эндокринол. (Лозанна) 5 , 6 (2014).
Артикул
Google Scholar
Трабадо, С. и др. .Инсулиноподобный пептид 3 (INSL3) у мужчин с врожденным гипогонадотропным гипогонадизмом / синдромом Каллмана и эффекты различных методов гормонального лечения: одноцентровое исследование с участием 281 пациента. J. Clin. Эндокринол. Метаб. 99 , E268 – E275 (2014).
Артикул
CAS
PubMed
Google Scholar
Нильсен, К. Т. и др. . Начало выделения сперматозоидов (сперматозоидов) у мальчиков в зависимости от возраста, роста яичек, волос на лобке и роста. J. Clin. Эндокринол. Метаб. 62 , 532–535 (1986).
Артикул
CAS
PubMed
Google Scholar
Биро, Ф. М. и др. . Начало развития груди в продольной когорте. Педиатрия 132 , 1019–1027 (2013).
Артикул
PubMed
PubMed Central
Google Scholar
Херман-Гидденс, М.Е. и др. . Вторичные половые признаки у мальчиков: данные Pediatric Research in Office Settings Network. Педиатрия 130 , e1058 – e1068 (2012).
Артикул
PubMed
Google Scholar
Аксглаэде, Л., Соренсен, К., Петерсен, Дж. Х., Скаккебек, Н. Э. и Юул, А. Недавнее снижение возраста груди: исследование полового созревания в Копенгагене. Педиатрия 123 , e932 – e939 (2009).
Артикул
PubMed
Google Scholar
Соренсен, К., Аксгладе, Л., Петерсен, Дж. Х. и Юул, А. Недавние изменения в сроках полового созревания у здоровых датских мальчиков: ассоциации с индексом массы тела. J. Clin. Эндокринол. Метаб. 95 , 263–270 (2010).
Артикул
CAS
PubMed
Google Scholar
Evain-Brion, D., Gendrel, D., Bozzola, M., Chaussain, J. L. & Job, J. C. Диагностика синдрома Каллмана в раннем младенчестве. Acta Paediatr. Сканд. 71 , 937–940 (1982).
Артикул
CAS
PubMed
Google Scholar
Грумбах, М. М. Окно возможностей: диагностика дефицита гонадотропинов у младенцев мужского пола. J. Clin. Эндокринол. Метаб. 90 , 3122–3127 (2005).
Артикул
CAS
PubMed
Google Scholar
Каплан Дж.Д., Бернштейн, Дж. А., Кван, А. и Хаджинс, Л. Ключи к ранней диагностике синдрома Каллмана. Am. J. Med. Genet. А 152А , 2796–2801 (2010).
Артикул
PubMed
Google Scholar
Майн, К. М., Шмидт, И. М. и Скаккебек, Н. Е. Возможная роль репродуктивных гормонов у новорожденных мальчиков: прогрессирующий гипогонадизм без послеродового пика тестостерона. J. Clin. Эндокринол. Метаб. 85 , 4905–4907 (2000).
Артикул
CAS
PubMed
Google Scholar
Wohlfahrt-Veje, C. и др. . Приобретенный крипторхизм часто встречается в младенчестве и детстве. Внутр. Дж. Андрол. 32 , 423–428 (2009).
Артикул
PubMed
Google Scholar
Baetens, D. et al. . Обширный клинический, гормональный и генетический скрининг в большой последовательной серии из 46 новорожденных XY и младенцев с атипичным половым развитием. Orphanet J. Rare Dis. 9 , 209 (2014).
Артикул
PubMed
PubMed Central
Google Scholar
Хатипоглу Н. и Куртоглу С. Микропенис: этиология, диагностика и подходы к лечению. J. Clin. Res. Педиатр. Эндокринол. 5 , 217–223 (2013).
Артикул
PubMed
PubMed Central
Google Scholar
Боас, М. и др. . Длина и скорость роста полового члена в послеродовом периоде коррелируют с уровнем тестостерона в сыворотке: продольное исследование 1962 нормальных мальчиков. евро. J. Endocrinol. 154 , 125–129 (2006).
Артикул
CAS
PubMed
Google Scholar
Томова А. и др. . Рост и развитие мужских наружных половых органов: поперечное исследование 6200 мужчин в возрасте от 0 до 19 лет. Arch. Педиатр.Adolesc. Med. 164 , 1152–1157 (2010).
Артикул
PubMed
Google Scholar
Юул А. и др. . Пубертатное развитие у датских детей: сравнение последних европейских и американских данных. Внутр. Дж. Андрол. 29 , 247–255 (2006).
Артикул
CAS
PubMed
Google Scholar
Палмерт, М. Р.& Дункель, Л. Клиническая практика. Задержка полового созревания. N. Engl. J. Med. 366 , 443–453 (2012).
Артикул
CAS
PubMed
Google Scholar
van Buuren, S. & Ooms, J. C. Этапная линейная диаграмма: условная справочная диаграмма для отслеживания развития. Stat. Med. 28 , 1569–1579 (2009).
Артикул
PubMed
Google Scholar
Питтелуд, Н. и др. . Роль предшествующего полового созревания, биохимических маркеров созревания яичек и генетики в выяснении фенотипической гетерогенности идиопатического гипогонадотропного гипогонадизма. J. Clin. Эндокринол. Метаб. 87 , 152–160 (2002).
Артикул
CAS
PubMed
Google Scholar
Шоу, Н. Д. и др. . Расширение фенотипа и генотипа дефицита женского гонадолиберина. J. Clin. Эндокринол. Метаб. 96 , E566 – E576 (2011).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Янг, Дж. Подход к пациенту мужского пола с врожденным гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 97 , 707–718 (2012).
Артикул
CAS
PubMed
Google Scholar
Лавэц, Дж.Г. и др. . Оценка 451 датского мальчика с задержкой полового созревания: диагностическое использование новой номограммы полового созревания и эффекты пероральной терапии тестостероном. J. Clin. Эндокринол. Метаб. 100 , 1376–1385 (2015).
Артикул
CAS
PubMed
Google Scholar
Герой, М. и др. . Детский рост самок с синдромом Каллмана и мутациями FGFR1 . Clin. Эндокринол.(Oxf.) 82 , 122–126 (2015).
Артикул
CAS
Google Scholar
Хаффер В., Скотт В. Х., Коннор Т. Б. и Ловис Х. Психологические исследования взрослых пациентов мужского пола с сексуальным инфантилизмом до и после терапии андрогенами. Ann. Междунар. Med. 61 , 255–268 (1964).
Артикул
CAS
PubMed
Google Scholar
Дуайер, А.А., Куинтон, Р., Морин, Д. и Питтелуд, Н. Определение неудовлетворенных потребностей в отношении здоровья пациентов с врожденным гипогонадотропным гипогонадизмом с использованием оценки потребностей через Интернет: значение для онлайн-вмешательств и поддержки со стороны коллег. Orphanet J. Rare Dis. 9 , 83 (2014).
Артикул
PubMed
PubMed Central
Google Scholar
Айдоган, У. и др. . Повышенная частота тревожности, депрессии, повышение качества жизни и сексуальной жизни у молодых гипогонадотропных гипогонадных мужчин и влияние заместительной терапии тестостероном на эти состояния. Endocr. J. 59 , 1099–1105 (2012).
Артикул
PubMed
Google Scholar
Lasaite, L., Ceponis, J., Preiksa, R. T. & Zilaitiene, B. Нарушение эмоционального состояния, качества жизни и когнитивных функций у молодых мужчин с гипогонадизмом. Андрология 46 , 1107–1112 (2014).
Артикул
CAS
PubMed
Google Scholar
Куинтон, Р. и др. . Идиопатический дефицит гонадотропина: генетические вопросы, решаемые посредством фенотипической характеристики. Clin. Эндокринол. (Oxf.) 55 , 163–174 (2001).
Артикул
CAS
Google Scholar
Левковиц-Шпунтов, Х. М. и др. . Обонятельный фенотипический спектр при идиопатическом гипогонадотропном гипогонадизме: патофизиологические и генетические последствия. J. Clin.Эндокринол. Метаб. 97 , E136 – E144 (2012).
Артикул
CAS
PubMed
Google Scholar
Делла Валле, Э. и др. . Распространенность обонятельных и других аномалий развития у пациентов с центральным гипогонадотропным гипогонадизмом. Фронт. Эндокринол. (Лозанна) 4 , 70 (2013).
Артикул
Google Scholar
Коста-Барбоса, Ф.А. и др. . Приоритет генетического тестирования у пациентов с синдромом Каллмана с использованием клинических фенотипов. J. Clin. Эндокринол. Метаб. 98 , E943 – E953 (2013).
Артикул
PubMed
PubMed Central
Google Scholar
Krams, M. et al. . Синдром Каллмана: зеркальные движения, связанные с двусторонней гипертрофией кортикоспинального тракта. Неврология 52 , 816–822 (1999).
Артикул
CAS
PubMed
Google Scholar
Лайтинен, Э. М. и др. . Заболеваемость, фенотипические особенности и молекулярная генетика синдрома Каллмана в Финляндии. Orphanet J. Rare Dis. 6 , 41 (2011).
Артикул
PubMed
PubMed Central
Google Scholar
Bailleul-Forestier, I. et al. .Агенез зубов у людей с синдромом Каллмана с мутациями FGFR1 . Внутр. J. Paediatr. Вмятина. 20 , 305–312 (2010).
Артикул
PubMed
Google Scholar
Molsted, K., Kjaer, I., Giwercman, A., Vesterhauge, S. & Skakkebaek, N. E. Морфология черепа у пациентов с синдромом Каллмана с расщелиной губы и неба и без нее. Черепная пасть неба. J. 34 , 417–424 (1997).
Артикул
CAS
PubMed
Google Scholar
Wehkalampi, K., Widen, E., Laine, T., Palotie, A. & Dunkel, L. Особенности наследования конституциональной задержки роста и полового созревания в семьях девочек и мальчиков подросткового возраста, направленных в специализированные педиатрические отделения забота. J. Clin. Эндокринол. Метаб. 93 , 723–728 (2008).
Артикул
CAS
PubMed
Google Scholar
Waldstreicher, J. и др. . Генетическая и клиническая гетерогенность дефицита гонадотропин-рилизинг-гормона у человека. J. Clin. Эндокринол. Метаб. 81 , 4388–4395 (1996).
CAS
PubMed
Google Scholar
Чжу Дж. и др. . Общая генетическая основа самоограничивающейся задержки полового созревания и идиопатического гипогонадотропного гипогонадизма. J. Clin. Эндокринол. Метаб. 100 , E646 – E654 (2015).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Франко Б. и др. . Ген, удаленный при синдроме Каллмана, имеет гомологию с адгезией нервных клеток и молекулами, определяющими путь аксонов. Nature 353 , 529–536 (1991).
Артикул
CAS
PubMed
Google Scholar
Legouis, R. et al. .Ген-кандидат для Х-сцепленного синдрома Каллмана кодирует белок, связанный с молекулами адгезии. Cell 67 , 423–435 (1991).
Артикул
CAS
PubMed
Google Scholar
Харделин, Дж. П. и др. . Синдром Каллмана, сцепленный с X-хромосомой: остановите мутации, чтобы подтвердить ген-кандидат. Proc. Natl Acad. Sci. США 89 , 8190–8194 (1992).
Артикул
CAS
PubMed
Google Scholar
Бик, Д. и др. . Краткое сообщение: внутригенная делеция гена KALIG-1 при синдроме Каллмана. N. Engl. J. Med. 326 , 1752–1755 (1992).
Артикул
CAS
PubMed
Google Scholar
Харделин, Дж. П. и др. . Аносмин-1 является регионально ограниченным компонентом базальных мембран и интерстициальных матриц во время органогенеза: последствия для аномалий развития синдрома Каллмана, сцепленного с Х-хромосомой. Dev. Дин. 215 , 26–44 (1999).
Артикул
CAS
PubMed
Google Scholar
Гонсалес-Мартинес, Д. и др. . Аносмин-1 модулирует передачу сигналов рецептора 1 фактора роста фибробластов в обонятельных нейробластах гонадотропин-рилизинг-гормона человека посредством гепарансульфат-зависимого механизма. J. Neurosci. 24 , 10384–10392 (2004).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Эндо, Ю., Ishiwata-Endo, H. & Yamada, K. M. Белок внеклеточного матрикса аносмин способствует формированию нервного гребня и регулирует активности FGF, BMP и WNT. Dev. Ячейка 23 , 305–316 (2012).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Трарбах, Э. Б. и др. . Нонсенс мутации в гене FGF8 , вызывающие различную степень дефицита высвобождения гонадотропина у человека. J. Clin. Эндокринол. Метаб. 95 , 3491–3496 (2010).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Сарфати Дж. и др. . Сравнительное фенотипическое исследование пациентов с синдромом Каллмана, несущих моноаллельные и двуаллельные мутации в генах прокинетицина 2 или рецептора прокинетицина 2. J. Clin. Эндокринол. Метаб. 95 , 659–669 (2010).
Артикул
CAS
PubMed
Google Scholar
Мартин, К. и др. . Роль пути прокинетицина 2 в воспроизводстве человека: данные исследования мутаций генов человека и мыши. Endocr. Ред. 32 , 225–246 (2011).
Артикул
CAS
PubMed
Google Scholar
де Ру, Н. и др. .Семья с гипогонадотропным гипогонадизмом и мутациями рецептора гонадотропин-рилизинг-гормона. N. Engl. J. Med. 337 , 1597–1602 (1997).
Артикул
CAS
PubMed
Google Scholar
Лайман, Л. К. и др. . Мутации в гене рецептора гонадотропин-рилизинг-гормона вызывают гипогонадотропный гипогонадизм. Nat. Genet. 18 , 14–15 (1998).
Артикул
CAS
PubMed
Google Scholar
Bouligand, J. и др. . Изолированный семейный гипогонадотропный гипогонадизм и мутация GNRh2 . N. Engl. J. Med. 360 , 2742–2748 (2009).
Артикул
CAS
PubMed
Google Scholar
Чан, Ю. М. и др. . GNRh2 мутации у пациентов с идиопатическим гипогонадотропным гипогонадизмом. Proc. Natl Acad. Sci. США 106 , 11703–11708 (2009).
Артикул
PubMed
Google Scholar
Чан, Ю. М. и др. . ГнРГ-дефицитные фенотипы у людей и мышей с гетерозиготными вариантами в KISS1 / Kiss1 . J. Clin. Эндокринол. Метаб. 96 , E1771 – E1781 (2011).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Au, М.Г., Кроули, В. Ф. Младший и Бак, К. Л. Генетическое консультирование при изолированном дефиците гонадолиберина. Мол. Клетка. Эндокринол. 346 , 102–109 (2011).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Питтелуд, Н. и др. . Дигенные мутации являются причиной вариабельных фенотипов при идиопатическом гипогонадотропном гипогонадизме. J. Clin. Вкладывать деньги. 117 , 457–463 (2007).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Перри, Дж. Р. и др. . Аллельные ассоциации, специфичные для родителей по происхождению, среди 106 геномных локусов для возраста менархе. Nature 514 , 92–97 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Андерссон, А. М. и др. .Продольные профили репродуктивных гормонов у младенцев: пиковые уровни ингибина B у мальчиков грудного возраста превышают уровни у взрослых мужчин. J. Clin. Эндокринол. Метаб. 83 , 675–681 (1998).
CAS
PubMed
Google Scholar
Chellakooty, M. и др. . Уровни ингибина A, ингибина B, фолликулостимулирующего гормона, лютеинизирующего гормона, эстрадиола и глобулина, связывающего половые гормоны, у 473 здоровых девочек-младенцев. J. Clin. Эндокринол. Метаб. 88 , 3515–3520 (2003).
Артикул
CAS
PubMed
Google Scholar
Waldstreicher, J. et al. . Генетическая и клиническая гетерогенность дефицита гонадотропин-рилизинг-гормона у человека. J. Clin. Эндокринол. Метаб. 81 , 4388–4395 (1996).
CAS
PubMed
Google Scholar
Маршалл, В.А. и Таннер, Дж. М. Вариации паттернов пубертатных изменений у девочек. Arch. Дис. Ребенок. 44 , 291–303 (1969).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Маршалл, В. А. и Таннер, Дж. М. Вариации в характере пубертатных изменений у мальчиков. Arch. Дис. Ребенок. 45 , 13–23 (1970).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Каллманн, Ф.Дж., Шенфельд, В. А. и Баррера, С. Е. Генетические аспекты первичного евнухоидизма. Am. J. Ment. Дефицит. 48 , 203–236 (1944).
Google Scholar
Segal, TY, Mehta, A., Anazodo, A., Hindmarsh, PC & Dattani, MT Роль гонадотропин-рилизинг-гормона и тестов стимуляции хорионического гонадотропина человека в дифференциации пациентов с гипогонадотропным гипогонадизмом от пациентов с конституциональной задержкой рост и половое созревание. J. Clin. Эндокринол. Метаб. 94 , 780–785 (2009).
Артикул
CAS
PubMed
Google Scholar
Биндер, Г., Швейцер, Р., Блюменшток, Г. и Браун, Р. Тест на ингибин В плюс ЛГ по сравнению с агонистом гонадолиберина для отличия конституциональной задержки роста и полового созревания от изолированного гипогонадотропного гипогонадизма у мальчиков. Clin. Эндокринол. (Oxf.) 82 , 100–105 (2015).
Артикул
CAS
Google Scholar
Кутан, Р. и др. . Исходные показатели ингибина B и антимюллерова гормона для диагностики гипогонадотропного гипогонадизма (ГГ) у мальчиков с задержкой полового созревания. J. Clin. Эндокринол. Метаб. 95 , 5225–5232 (2010).
Артикул
CAS
PubMed
Google Scholar
Адан Л. и др. . Концентрация ингибина B в плазме и антимуллеровых гормонов у мальчиков: различение между врожденным гипогонадотропным гипогонадизмом и конституциональной задержкой полового созревания. Med. Sci. Монит. 16 , CR511 – CR517 (2010).
CAS
PubMed
Google Scholar
Янг, Дж. и др. . Антимуллеровый гормон у пациентов с гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 84 , 2696–2699 (1999).
CAS
PubMed
Google Scholar
Vizeneux, A. et al. .Врожденный гипогонадотропный гипогонадизм в детстве: клинические проявления и генетический анализ у 46 мальчиков. PLoS ONE 8 , e77827 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Бэй, К. и др. . Уровни инсулиноподобного фактора 3 в сыворотке у 135 нормальных мужчин и 85 мужчин с заболеваниями яичек: связь с осью лютеинизирующий гормон-тестостерон. Дж.Clin. Эндокринол. Метаб. 90 , 3410–3418 (2005).
Артикул
CAS
PubMed
Google Scholar
Харрингтон Дж. И Палмерт М. Р. Клинический обзор: Отличие конституциональной задержки роста и полового созревания от изолированного гипогонадотропного гипогонадизма: критическая оценка доступных диагностических тестов. J. Clin. Эндокринол. Метаб. 97 , 3056–3067 (2012).
Артикул
CAS
PubMed
Google Scholar
Йохансен, М.Л. и др. . Сывороточные уровни инсулиноподобного фактора 3, антимюллеровского гормона, ингибина B и тестостерона во время полового созревания у здоровых мальчиков: продольное пилотное исследование. Репродукция 147 , 529–535 (2014).
Артикул
CAS
PubMed
Google Scholar
Чан, Ю. М. и др. . Введение экзогенного кисспептина в качестве зонда функции нейронов ГнРГ у пациентов с идиопатическим гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 99 , E2762 – E2771 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Янг, Дж. и др. . Кисспептин восстанавливает пульсирующую секрецию ЛГ у пациентов с дефицитом передачи сигналов нейрокинина B: физиологические, патофизиологические и терапевтические последствия. Нейроэндокринология 97 , 193–202 (2013).
Артикул
CAS
PubMed
Google Scholar
Джаясена, К.Н. и др. . Прием кисспептина-54 два раза в неделю в течение 8 недель стимулирует высвобождение репродуктивных гормонов у женщин с гипоталамической аменореей. Clin. Pharmacol. Ther. 88 , 840–847 (2010).
Артикул
CAS
PubMed
Google Scholar
Джаясена, К. Н. и др. . Кисспептин-54 запускает созревание яйцеклеток у женщин, подвергшихся оплодотворению in vitro . J. Clin. Вкладывать деньги. 124 , 3667–3677 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Сикиотис, Г. П. и др. . Врожденный идиопатический гипогонадотропный гипогонадизм: свидетельство дефектов гипоталамуса, гипофиза и яичек. J. Clin. Эндокринол. Метаб. 95 , 3019–3027 (2010).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Харделин, Дж.P. и др. . Неоднородность мутаций, ответственных за синдром Каллмана, сцепленный с Х-хромосомой. Hum. Мол. Genet. 2 , 373–377 (1993).
Артикул
CAS
PubMed
Google Scholar
Сюй Н. и др. . Мутация в гене рецептора 1 фактора роста фибробластов вызывает полностью проникающий нормосмический изолированный гипогонадотропный гипогонадизм. J. Clin. Эндокринол. Метаб. 92 , 1155–1158 (2007).
Артикул
CAS
PubMed
Google Scholar
Питтелуд, Н. и др. . Обратимый синдром Каллмана, задержка полового созревания и изолированная аносмия, встречающиеся в одной семье с мутацией в гене рецептора 1 фактора роста фибробластов. J. Clin. Эндокринол. Метаб. 90 , 1317–1322 (2005).
Артикул
CAS
PubMed
Google Scholar
Мирауи, Х., Dwyer, A. & Pitteloud, N. Роль передачи сигналов фактора роста фибробластов (FGF) в нейроэндокринном контроле репродукции человека. Мол. Клетка. Эндокринол. 346 , 37–43 (2011).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Кирк, Дж. М. и др. . Односторонняя аплазия почек при Х-сцепленном синдроме Каллмана. Clin. Genet. 46 , 260–262 (1994).
Артикул
CAS
PubMed
Google Scholar
Георгопулос, Н. А. и др. . Дисгенезия почек и дефекты гена KAL1 у пациентов со спорадическим синдромом Каллмана. Fertil. Стерил. 88 , 1311–1317 (2007).
Артикул
CAS
PubMed
Google Scholar
Джексон Р. С. и др. .Ожирение и нарушение обработки прогормона, связанные с мутациями гена прогормон-конвертазы 1 человека. Nat. Genet. 16 , 303–306 (1997).
Артикул
CAS
PubMed
Google Scholar
Вильянуэва, К. и др. . Врожденный гипогонадотропный гипогонадизм с расщепленным пороком развития кисти и стопы: клиническое заболевание с высокой частотой мутаций FGFR1 . Genet.Med. http://dx.doi.org/10.1038/gim.2014.166.
Ballabio, A. и др. . Делеции гена стероидной сульфатазы при «классическом» Х-сцепленном ихтиозе и при Х-сцепленном ихтиозе, связанном с синдромом Каллмана. Hum. Genet. 77 , 338–341 (1987).
Артикул
CAS
PubMed
Google Scholar
Vermeulen, S. и др. . Синдром Каллмана у пациента с врожденным сфероцитозом и интерстициальным 8p11.2 удаления. Am. J. Med. Genet. 108 , 315–318 (2002).
Артикул
PubMed
Google Scholar
Баллабио, А. и Андрия, Г. Делеции и транслокации, вовлекающие дистальное короткое плечо Х-хромосомы человека: обзор и гипотезы. Hum. Мол. Genet. 1 , 221–227 (1992).
Артикул
CAS
PubMed
Google Scholar
Вассон, А. и др. . CGH на основе настраиваемого олигонуклеотидного массива: надежный диагностический инструмент для обнаружения изменений экзонного числа копий в нескольких генах-мишенях. евро. J. Hum. Genet. 21 , 977–987 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Доде, К. и Харделин, Дж. П. Клиническая генетика синдрома Каллмана. Ann. Эндокринол. (Париж) 71 , 149–157 (2010).
Артикул
CAS
Google Scholar
Лу, Дж. Т., Кампо, П. М. и Ли, Б. Х. Корреляция генотип-фенотип — беспорядочные половые связи в эпоху секвенирования следующего поколения. N. Engl. J. Med. 371 , 593–596 (2014).
Артикул
PubMed
Google Scholar
Ли, П. А. и др. . Отцовство после одностороннего крипторхизма: контролируемое исследование. Педиатрия 98 , 676–679 (1996).
CAS
PubMed
Google Scholar
ван Бракель, Дж. и др. . Потенциал фертильности у мужчин с врожденными неопущенными яичками в анамнезе: долгосрочное катамнестическое исследование. Андрология 1 , 100–108 (2013).
Артикул
CAS
PubMed
Google Scholar
Ритцен, Э.М. и др. . Северный консенсус относительно лечения неопущенных яичек. Acta Paediatr. 96 , 638–643 (2007).
Артикул
PubMed
Google Scholar
Пенсон Д., Кришнасвами С., Джулс А. и Макфитерс М. Л. Эффективность гормональной и хирургической терапии крипторхизма: систематический обзор. Педиатрия 131 , e1897 – e1907 (2013).
Артикул
PubMed
PubMed Central
Google Scholar
Чан, Э., Wayne, C., Nasr, A. & FRCSC для Канадской ассоциации детских хирургов, основанных на фактических данных. Идеальное время орхиопексии: систематический обзор. Pediatr. Surg. Int. 30 , 87–97 (2014).
Артикул
PubMed
Google Scholar
Бин-Аббас, Б., Конте, Ф. А., Грумбах, М. М. и Каплан, С. Л. Врожденный гипогонадотропный гипогонадизм и микропенис: влияние лечения тестостероном на размер полового члена у взрослых, почему изменение пола не показано. J. Pediatr. 134 , 579–583 (1999).
Артикул
CAS
PubMed
Google Scholar
Майн, К. М., Шмидт, И. М., Топпари, Дж. И Скаккебек, Н. Е. Раннее постнатальное лечение гипогонадотропного гипогонадизма рекомбинантными человеческими ФСГ и ЛГ. евро. J. Endocrinol. 146 , 75–79 (2002).
Артикул
CAS
PubMed
Google Scholar
Буваттье, К. и др. . Неонатальная терапия гонадотропинами при мужском врожденном гипогонадотропном гипогонадизме. Nat. Rev. Endocrinol. 8 , 172–182 (2012).
Артикул
CAS
Google Scholar
Chemes, H.E. и др. . Физиологическая нечувствительность к андрогенам яичек плода, новорожденного и раннего детства объясняется онтогенезом экспрессии рецепторов андрогенов в клетках Сертоли. Дж.Clin. Эндокринол. Метаб. 93 , 4408–4412 (2008).
Артикул
CAS
PubMed
Google Scholar
Рей, Р. А., Мюсс, М., Венара, М. и Чемес, Х. Э. Онтогенез экспрессии рецепторов андрогенов в плодных и постнатальных семенниках: его значение для созревания клеток Сертоли и начала сперматогенеза взрослых. Microsc. Res. Tech. 72 , 787–795 (2009).
Артикул
CAS
PubMed
Google Scholar
Дункель, Л.& Куинтон, Р. Переход в эндокринологии: индукция полового созревания. евро. J. Endocrinol. 170 , R229 – R239 (2014).
Артикул
CAS
PubMed
Google Scholar
Дуайер, А. А., Фан-Хуг, Ф., Хаушильд, М., Элоу-Груау, Э. и Питтелуд, Н. Переход в эндокринологии: гипогонадизм в подростковом возрасте. евро. J. Endocrinol. 173 , R15 – R24 (2015).
Артикул
CAS
PubMed
Google Scholar
Бобров, Н.А., Мани, Дж. И Льюис, В. Г. Задержка полового созревания, эротизм и обоняние: психологическое исследование гипогонадотропизма, осматического и аносматического (синдром Каллмана). Arch. Секс. Behav. 1 , 329–344 (1971).
Артикул
CAS
PubMed
Google Scholar
Хан, Т. С. и Булу, П. М. Какая оптимальная терапия для молодых мужчин с гипогонадотропным гипогонадизмом? Clin. Эндокринол. 72 , 731–737 (2010).
Артикул
CAS
Google Scholar
Santhakumar, A., Miller, M. & Quinton, R. Индукция полового созревания у взрослых мужчин с изолированным гипогонадотропным гипогонадизмом с использованием пролонгированного внутримышечного 1-г депо тестостерона ундеканоата (Небидо). Clin. Эндокринол. (Oxf.) 80 , 155–157 (2014).
Артикул
CAS
Google Scholar
Куинтон, Р. и др. . Синдром Каллмана: всегда ли он на всю жизнь? Clin. Эндокринол. (Oxf.) 50 , 481–485 (1999).
Артикул
CAS
Google Scholar
Синиси А. А. и др. . Гомозиготная мутация в гене прокинетицин-рецептора 2 (Val274Asp), проявляющаяся в виде обратимого синдрома Каллмана и стойкой олигозооспермии: клинический случай. Hum. Репродукция. 23 , 2380–2384 (2008).
Артикул
CAS
PubMed
Google Scholar
Tommiska, J., Jorgensen, N., Christiansen, P., Juul, A. & Raivio, T. Гомозиготная мутация R262Q в рецепторе гонадотропин-рилизинг гормона, проявляющаяся как реверсирование гипогонадотропного гипогонадизма с поздним началом гипогонадизм. Clin. Эндокринол. (Oxf.) 78 , 316–317 (2013).
Артикул
CAS
Google Scholar
Лайтинен, Э.М. и др. . Обратимый врожденный гипогонадотропный гипогонадизм у пациентов с мутациями CHD7 , FGFR1 или GNRHR . PLoS ONE 7 , e39450 (2012).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Кульшрешта, Б., Хадгават, Р., Гупта, Н. и Аммини, А. Прогрессирование полового созревания после начала терапии андрогенами у пациентов с идиопатическим гипогонадотропным гипогонадизмом. Indian J. Endocrinol. Метаб. 17 , 851–854 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Santhakumar, A., Balasubramanian, R., Miller, M. & Quinton, R. Обращение изолированного гипогонадотропного гипогонадизма: долговременная целостность оси гипоталамо-гипофиз-яички у двух мужчин зависит от периодического воздействия андрогенов . Clin. Эндокринол. (Oxf.) 81 , 473–476 (2014).
Артикул
CAS
Google Scholar
Мао, Дж. Ф. и др. . Обращение идиопатического гипогонадотропного гипогонадизма: когортное исследование у китайских пациентов. Азиатский Дж. Андрол. 17 , 497–502 (2014).
PubMed Central
Google Scholar
Delemarre-Van de Waal, H. A. Индукция роста яичек и сперматогенеза пульсирующим внутривенным введением гонадотропин-рилизинг-гормона пациентам с гипогонадотропным гипогонадизмом. Clin. Эндокринол. (Oxf.) 38 , 473–480 (1993).
Артикул
CAS
Google Scholar
Hoffman, A. R. & Crowley, W. F. Jr. Индукция полового созревания у мужчин путем длительного пульсирующего введения низких доз гонадотропин-рилизинг-гормона. N. Engl. J. Med. 307 , 1237–1241 (1982).
Артикул
CAS
PubMed
Google Scholar
Гонг, К., Лю Ю., Цинь М., Ву Д. и Ван X. Пульсирующий ГнРГ превосходит ХГЧ по терапевтической эффективности у мальчиков-подростков с гипогонадотропным гипогонадодизмом. J. Clin. Эндокринол. Метаб. http://dx.doi.org/10.1210/jc.2015-1343.
Bouvattier, C., Tauber, M., Jouret, B., Chaussain, J. L. & Rochiccioli, P. Лечение гонадотропинами гипогонадотропных гипогонадных подростков. J. Pediatr. Эндокринол. Метаб. 12 (Дополнение 1), 339–344 (1999).
PubMed
Google Scholar
Баррио, Р., де Луис, Д., Алонсо, М., Ламас, А. и Морено, Дж. С. Индукция полового созревания с помощью хорионического гонадотропина человека и фолликулостимулирующего гормона у мужчин-подростков с гипогонадотропным гипогонадизмом. Fertil. Стерил. 71 , 244–248 (1999).
Артикул
CAS
PubMed
Google Scholar
Сираиси, К., Ока, С. & Мацуяма, Х. Оценка качества жизни во время лечения гонадотропинами мужского гипогонадотропного гипогонадизма. Clin. Эндокринол. (Oxf.) 81 , 259–265 (2014).
Артикул
CAS
Google Scholar
Варимо, Т., Герой, М., Лайтинен, Э. М., Синтонен, Х. и Райвио, Т. Связанное со здоровьем качество жизни пациентов мужского пола с врожденным гипогонадотропным гипогонадизмом. Clin. Эндокринол.(Oxf.) http://dx.doi.org/10.1111/cen.12701.
Питтелуд, Н. и др. . Предикторы исхода длительной терапии ГнРГ у мужчин с идиопатическим гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 87 , 4128–4136 (2002).
Артикул
CAS
PubMed
Google Scholar
Беррис, А. С., Родбард, Х. У., Винтерс, С. Дж. И Шеринс, Р. Дж. Терапия гонадотропинами у мужчин с изолированным гипогонадотропным гипогонадизмом: реакция на хорионический гонадотропин человека определяется начальным размером яичек. J. Clin. Эндокринол. Метаб. 66 , 1144–1151 (1988).
Артикул
CAS
PubMed
Google Scholar
Миягава, Ю. и др. . Результат терапии гонадотропинами мужского гипогонадотропного гипогонадизма в университетских центрах мужского бесплодия: 30-летнее ретроспективное исследование. J. Urol. 173 , 2072–2075 (2005).
Артикул
CAS
PubMed
Google Scholar
Лю П.Ю. и др. . Индукция сперматогенеза и фертильности во время лечения гонадотропином мужчин с гонадотропным дефицитом бесплодия: предикторы исхода фертильности. J. Clin. Эндокринол. Метаб. 94 , 801–808 (2009).
Артикул
CAS
PubMed
Google Scholar
Warne, D. W. и др. . Комбинированный анализ данных для определения прогностических факторов сперматогенеза у мужчин с гипогонадотропным гипогонадизмом, получавших рекомбинантный фолликулостимулирующий гормон человека и хорионический гонадотропин человека. Fertil. Стерил. 92 , 594–604 (2009).
Артикул
CAS
PubMed
Google Scholar
Дуайер, А. А., Райвио, Т. и Питтелуд, Н. Замена гонадотропинов для индукции фертильности у мужчин с гипогонадизмом. Best Pract. Res. Clin. Эндокринол. Метаб. 29 , 91–103 (2015).
Артикул
CAS
PubMed
Google Scholar
Бухтер, Д., Behre, H. M., Kliesch, S. & Nieschlag, E. Pulsatile GnRH или человеческий хорионический гонадотропин / человеческий менопаузальный гонадотропин как эффективное лечение для мужчин с гипогонадотропным гипогонадизмом: обзор 42 случаев. евро. J. Endocrinol. 139 , 298–303 (1998).
Артикул
CAS
PubMed
Google Scholar
Растрелли, Дж., Корона, Дж., Маннуччи, Э. и Магги, М. Факторы, влияющие на сперматогенез при заместительной гонадотропиновой терапии: метааналитическое исследование. Андрология 2 , 794–808 (2014).
Артикул
CAS
PubMed
Google Scholar
Schopohl, J., Mehltretter, G., von Zumbusch, R., Eversmann, T. и von Werder, K. Сравнение гонадотропин-рилизинг-гормона и терапии гонадотропинами у пациентов мужского пола с идиопатическим гипоталамическим гипогонадизмом. Fertil. Стерил. 56 , 1143–1150 (1991).
Артикул
CAS
PubMed
Google Scholar
Лю Л., Бэнкс, С. М., Барнс, К. М. и Шеринс, Р. Дж. Двухлетнее сравнение реакций яичек на пульсирующий гонадотропин-рилизинг-гормон и экзогенные гонадотропины с самого начала терапии у мужчин с изолированным гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 67 , 1140–1145 (1988).
Артикул
CAS
PubMed
Google Scholar
Кирк, Дж. М., Сэвидж, М. О., Грант, Д. Б., Булу, П.М. и Бессер, Г. М. Функция гонад и ответ на терапию хорионическими и менопаузальными гонадотропинами у пациентов мужского пола с идиопатическим гипогонадотропным гипогонадизмом. Clin. Эндокринол. (Oxf.) 41 , 57–63 (1994).
Артикул
CAS
Google Scholar
Булу, П. М. и др. . Индукция сперматогенеза рекомбинантным фолликулостимулирующим гормоном (пурегон) у мужчин с гипогонадотропной азооспермией, которые не реагировали только на хорионический гонадотропин человека. Дж. Андрол. 24 , 604–611 (2003).
Артикул
CAS
PubMed
Google Scholar
Беррис, А. С., Кларк, Р. В., Вантман, Д. Дж. И Шеринс, Р. Дж. Низкая концентрация сперматозоидов не препятствует фертильности у мужчин с изолированным гипогонадотропным гипогонадизмом после терапии гонадотропинами. Fertil. Стерил. 50 , 343–347 (1988).
Артикул
CAS
PubMed
Google Scholar
Финкель, Д.М., Филлипс, Дж. Л. и Снайдер, П. Дж. Стимуляция сперматогенеза гонадотропинами у мужчин с гипогонадотропным гипогонадизмом. N. Engl. J. Med. 313 , 651–655 (1985).
Артикул
CAS
PubMed
Google Scholar
Викари, Э. и др. . Терапия только хорионическим гонадотропином человека индуцирует сперматогенез у мужчин с изолированным гипогонадотропным гипогонадизмом — долгосрочное наблюдение. Внутр. Дж. Андрол. 15 , 320–329 (1992).
Артикул
CAS
PubMed
Google Scholar
Kung, A. W., Zhong, Y. Y., Lam, K. S. & Wang, C. Индукция сперматогенеза гонадотропинами у китайских мужчин с гипогонадотропным гипогонадизмом. Внутр. Дж. Андрол. 17 , 241–247 (1994).
Артикул
CAS
PubMed
Google Scholar
Райвио Т., Toppari, J., Perheentupa, A., McNeilly, A. S. & Dunkel, L. Лечение мальчиков с дефицитом гонадотропина в препубертатном периоде рекомбинантным фолликулостимулирующим гормоном человека. Ланцет 350 , 263–264 (1997).
Артикул
CAS
PubMed
Google Scholar
Райвио Т., Викстром А. М. и Дункель Л. Лечение мальчиков с дефицитом гонадотропина рекомбинантным человеческим ФСГ: долгосрочное наблюдение и результат. евро. J. Endocrinol. 156 , 105–111 (2007).
Артикул
CAS
PubMed
Google Scholar
Йонг, Э. Л., Ли, К. О., Нг, С. С. и Ратнам, С. С. Индукция сперматогенеза при изолированном гипогонадотропном гипогонадизме с помощью гонадотропинов и раннее вмешательство с помощью интрацитоплазматической инъекции сперматозоидов. Hum. Репродукция. 12 , 1230–1232 (1997).
Артикул
CAS
PubMed
Google Scholar
Фахми, И. и др. . ИКСИ с использованием спермы яичек при мужском гипогонадотропном гипогонадизме, не отвечающем на терапию гонадотропинами. Hum. Репродукция. 19 , 1558–1561 (2004).
Артикул
CAS
PubMed
Google Scholar
Цорн, Б., Пфайфер, М., Вирант-Клун, И. и Меден-Вртовец, Х. Внутрицитоплазматическая инъекция сперматозоидов в дополнение к лечению гонадотропинами у бесплодных мужчин с гипогонадотропным гипогонадизмом. Внутр. Дж. Андрол. 28 , 202–207 (2005).
Артикул
PubMed
Google Scholar
Бакирчиоглу, М. Э., Эрден, Х. Ф., Чирай, Х. Н., Баязит, Н. и Бахчечи, М. Терапия гонадотропинами в сочетании с ИКСИ у мужчин с гипогонадотропным гипогонадизмом. Репродукция. Биомед. Интернет 15 , 156–160 (2007).
Артикул
CAS
PubMed
Google Scholar
Ресорлу, Б., Абдулмаджед, М. И., Кара, К., Унсал, А. и Айдос, К. Важна ли интрацитоплазматическая инъекция спермы для лечения гипогонадотропного гипогонадизма? Сравнение идиопатического и вторичного гипогонадотропного гипогонадизма. Hum. Fertil. (Камб.) 12 , 204–208 (2009).
Артикул
CAS
Google Scholar
Крабчи, К. и др. . Оценка качества индуцированного сперматогенеза у гипогонадотропных гипогонадических мужчин, получавших гонадотропины. Репродукция. Биомед. Интернет 22 , 277–283 (2011).
Артикул
CAS
PubMed
Google Scholar
Анкарберг-Линдгрен, К., Кристром, Б. и Норджаваара, Э. Физиологическая заместительная терапия эстрогенами для индукции полового созревания у девочек: клиническое обсервационное исследование. Horm. Res. Педиатр. 81 , 239–244 (2014).
Артикул
CAS
PubMed
Google Scholar
[Авторы не указаны] Рекомбинантный лютеинизирующий гормон человека (ЛГ) для поддержки развития фолликулов, индуцированного рекомбинантным фолликулостимулирующим гормоном (ФСГ), у ановуляторных женщин с дефицитом ЛГ и ФСГ: исследование по подбору дозы.Европейская группа по изучению рекомбинантного ЛГ человека. J. Clin. Эндокринол. Метаб. 83 , 1507–1514 (1998).
Deubzer, B., Weber, K., Lawrenz, B., Schweizer, R. & Binder, G. Дефицит антимюллерова гормона у девочек с врожденной недостаточностью множественных гормонов гипофиза. J. Clin. Эндокринол. Метаб. 99 , E1045 – E1049 (2014).
Артикул
CAS
PubMed
Google Scholar
Анавальт, Б.D. Подход к мужскому бесплодию и индукции сперматогенеза. J. Clin. Эндокринол. Метаб. 98 , 3532–3542 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Кулкарни, А. Д. и др. . Лечение бесплодия и многоплодие в США. N. Engl. J. Med. 369 , 2218–2225 (2013).
Артикул
CAS
PubMed
Google Scholar
Силвейра, Л.Ф. и Латронико А. С. Подход к пациенту с гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 98 , 1781–1788 (2013).
Артикул
CAS
PubMed
Google Scholar
Абель Б. С. и др. . Чувствительность к физиологическому режиму терапии ГнРГ и связь с генотипом у женщин с изолированным гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 98 , E206 – E216 (2013).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Ли, Р. Х. и Нг, Э. Х. Ведение ановуляторного бесплодия. Best Pract. Res. Clin. Акушерство. Gynaecol. 26 , 757–768 (2012).
Артикул
PubMed
Google Scholar
Шохам, З. и др. .Рекомбинантный ЛГ (лутропин альфа) для лечения гипогонадотропных женщин с выраженным дефицитом ЛГ: рандомизированное двойное слепое плацебо-контролируемое исследование с доказательством эффективности. Clin. Эндокринол. (Oxf.) 69 , 471–478 (2008).
Артикул
CAS
Google Scholar
Couzinet, B., Lestrat, N., Brailly, S., Forest, M. & Schaison, G. Стимуляция созревания фолликулов яичников чистым фолликулостимулирующим гормоном у женщин с дефицитом гонадотропина. J. Clin. Эндокринол. Метаб. 66 , 552–556 (1988).
Артикул
CAS
PubMed
Google Scholar
Шут, Д. К. и др. . Рекомбинантный фолликулостимулирующий гормон человека и реакция яичников у женщин с дефицитом гонадотропина. Hum. Репродукция. 9 , 1237–1242 (1994).
Артикул
CAS
PubMed
Google Scholar
Кауфманн, Р. и др. . Рекомбинантный лютеинизирующий гормон человека, лутропин альфа, для индукции развития фолликулов и беременности у женщин с серьезным дефицитом гонадотропина. Clin. Эндокринол. (Oxf.) 67 , 563–569 (2007).
CAS
Google Scholar
Мессини, И. Э. Индукция овуляции: мини-обзор. Hum. Репродукция. 20 , 2688–2697 (2005).
Артикул
CAS
PubMed
Google Scholar
Кван, И., Бхаттачарья, С., Канг, А. и Вулнер, А. Мониторинг стимулированных циклов при вспомогательной репродукции (ЭКО и ИКСИ). Кокрановская база данных систематических обзоров, , выпуск 8, ст. №: CD005289 http://dx.doi.org/10.1002/14651858.CD005289.pub3.
Семинара, С. Б. и др. . Успешное использование пульсирующего гонадотропин-рилизинг-гормона (GnRH) для индукции овуляции и беременности у пациентки с мутациями рецептора GnRH. J. Clin. Эндокринол. Метаб. 85 , 556–562 (2000).
CAS
PubMed
Google Scholar
Crowley, W. F. Jr & McArthur, J. W. Моделирование нормального менструального цикла при синдроме Каллмана путем пульсирующего введения высвобождающего гормона лютеинизирующего гормона (LHRH). J. Clin. Эндокринол. Метаб. 51 , 173–175 (1980).
Артикул
PubMed
Google Scholar
Санторо, Н., Filicori, M. & Crowley, W. F. Jr. Гипогонадотропные расстройства у мужчин и женщин: диагностика и терапия пульсирующим гонадотропин-рилизинг-гормоном. Endocr. Rev. 7 , 11–23 (1986).
Артикул
CAS
PubMed
Google Scholar
Ван, К. и др. . Длительное лечение гелем тестостерона (AndroGel) поддерживает положительное влияние на сексуальную функцию и настроение, мышечную и жировую массу, а также минеральную плотность костей у мужчин с гипогонадизмом. J. Clin. Эндокринол. Метаб. 89 , 2085–2098 (2004).
Артикул
CAS
PubMed
Google Scholar
Финкельштейн, Дж. С. и др. . Остеопороз у мужчин с идиопатическим гипогонадотропным гипогонадизмом. Ann. Междунар. Med. 106 , 354–361 (1987).
Артикул
CAS
PubMed
Google Scholar
Финкельштейн, Дж.С. и др. . Повышение плотности костей при лечении мужчин с идиопатическим гипогонадотропным гипогонадизмом. J. Clin. Эндокринол. Метаб. 69 , 776–783 (1989).
Артикул
CAS
PubMed
Google Scholar
Бер, Х. М., Клиш, С., Лейфке, Э., Линк, Т. М., Нишлаг, Э. Долгосрочное влияние терапии тестостероном на минеральную плотность костей у мужчин с гипогонадизмом. J. Clin.Эндокринол. Метаб. 82 , 2386–2390 (1997).
Артикул
CAS
PubMed
Google Scholar
Laitinen, E. M., Hero, M., Vaaralahti, K., Tommiska, J. & Raivio, T. Минеральная плотность костной ткани, состав тела и метаболизм костной ткани у пациентов с врожденным гипогонадотропным гипогонадизмом. Внутр. Дж. Андрол. 35 , 534–540 (2012).
Артикул
CAS
PubMed
Google Scholar
Хаяси, М. и др. . Остеопротекция семафорином 3А. Nature 485 , 69–74 (2012).
Артикул
CAS
PubMed
Google Scholar
Коли, Дж. А., Эль-Хадж Фулейхан, Г., Лаки, М. М. и FRAX ® Участники конференции по развитию позиции. FRAX ® Международная рабочая группа Объединенного международного общества клинической денситометрии и Международной конференции по разработке позиции Международного фонда остеопороза, 2010 г. J. Clin. Денситом. 14 , 237–239 (2011).
Артикул
PubMed
Google Scholar
Дин, Э. Л., Сонг, Ю., Малик, В. С. и Лю, С. Половые различия эндогенных половых гормонов и риск диабета 2 типа: систематический обзор и метаанализ. JAMA 295 , 1288–1299 (2006).
Артикул
CAS
PubMed
Google Scholar
Корона, г. и др. . Сахарный диабет 2 типа и тестостерон: исследование метаанализа. Внутр. Дж. Андрол. 34 , 528–540 (2011).
Артикул
CAS
PubMed
Google Scholar
Brand, JS, van der Tweel, I., Grobbee, DE, Emmelot-Vonk, MH & van der Schouw, YT Тестостерон, глобулин, связывающий половые гормоны, и метаболический синдром: систематический обзор и метаанализ наблюдательных исследований. Внутр. J. Epidemiol. 40 , 189–207 (2011).
Артикул
PubMed
Google Scholar
Бранд, Дж. С. и др. . Тестостерон, глобулин, связывающий половые гормоны, и метаболический синдром у мужчин: метаанализ данных отдельных участников обсервационных исследований. PLoS ONE 9 , e100409 (2014).
Артикул
CAS
PubMed
PubMed Central
Google Scholar
Зароцкий, В. и др. . Систематический обзор литературы о факторах риска, сопутствующих заболеваниях и последствиях гипогонадизма у мужчин. Андрология 2 , 819–834 (2014).
Артикул
CAS
PubMed
Google Scholar
Яламас, М. А. и др. . Острая отмена половых стероидов снижает чувствительность к инсулину у здоровых мужчин с идиопатическим гипогонадотропным гипогонадизмом. J. Clin. Эндокринол.Метаб. 92 , 4254–4259 (2007).
Артикул
CAS
PubMed
Google Scholar
Пугеат, М. и Николино, М. От педиатрической к эндокринологической помощи взрослым: проблема переходного периода. Pediatr. Эндокринол. Ред. 6 (Дополнение 4), 519–522 (2009).
PubMed
Google Scholar
Годбаут, А., Tejedor, I., Malivoir, S., Polak, M. & Touraine, P. Переход от педиатрического здравоохранения к медицинскому обслуживанию взрослых: оценка конкретных потребностей пациентов с хроническими эндокринными состояниями. Horm. Res. Педиатр. 78 , 247–255 (2012).
Артикул
CAS
PubMed
Google Scholar
Крауч, Н. С. и Крейтон, С. М. Переход к лечению подростков с нарушениями полового развития. Nat. Ред.Эндокринол. 10 , 436–442 (2014).
Артикул
PubMed
Google Scholar
В чем разница в патофизиологии гипергонадотропного гипогонадизма и гипогонадотропного гипогонадизма?
Автор
Мария Г. Вогиаци, доктор медицины Адъюнкт-профессор педиатрии, клинический педагог, Медицинская школа Перельмана при Пенсильванском университете; Директор Центра надпочечников и полового созревания, Отделение эндокринологии и диабета, Детская больница Филадельфии
Мария Г. Вогиаци, доктор медицинских наук, является членом следующих медицинских обществ: Общество эндокринологов, Общество педиатрической эндокринологии
Раскрытие информации: раскрывать нечего.
Специальная редакционная коллегия
Мэри Л. Виндл, PharmD Адъюнкт-профессор, Фармацевтический колледж Медицинского центра Университета Небраски; Главный редактор Medscape Drug Reference
Раскрытие информации: нечего раскрывать.
Барри Б. Берку, доктор медицины Профессор кафедры педиатрии, молекулярной фармакологии и физиологии, Медицинский колледж Университета Южной Флориды, Детская больница
Барри Б. Берку, доктор медицины, является членом следующих медицинских обществ: Американской академии педиатрии , Американская ассоциация клинических эндокринологов, Американская медицинская ассоциация, Американское педиатрическое общество, Ассоциация клинических ученых, Эндокринное общество, Медицинская ассоциация Флориды, Детское эндокринное общество, Общество педиатрических исследований, Южное общество педиатрических исследований, Общество изучения репродукции, американское Федерация клинических исследований, Гипофизарное общество
Раскрытие: Ничего не разглашать.
Главный редактор
Джордж Т. Гриффинг, доктор медицины Почетный профессор медицины, Медицинский факультет Университета Сент-Луиса
Джордж Т. Гриффинг, доктор медицины, является членом следующих медицинских обществ: Американской ассоциации содействия развитию науки, Международного общества клинической денситометрии, Южного Общество клинических исследований, Американский колледж руководителей медицинской практики, Американская ассоциация руководителей врачей, Американский колледж врачей, Американская диабетическая ассоциация, Американская федерация медицинских исследований, Американская кардиологическая ассоциация, Центральное общество клинических и трансляционных исследований, Эндокринное общество
Раскрытие информации : Нечего раскрывать.
Дополнительные участники
Брюс Бюлер, доктор медицины Профессор кафедры педиатрии и генетики, директор RSA, Медицинский центр Университета Небраски
Брюс Бюлер, доктор медицины, является членом следующих медицинских обществ: Американской академии церебрального паралича и медицины развития, Американской академии наук. Педиатрия, Американская ассоциация по вопросам интеллектуальных нарушений и нарушений развития, Американский колледж медицинской генетики и геномики, Американская ассоциация врачей-лидеров, Американская медицинская ассоциация, Медицинская ассоциация Небраски
Раскрытие: Ничего не разглашать.
Филлис В. Спайзер, доктор медицины Руководитель отделения детской эндокринологии Детского медицинского центра Стивена и Александры Коэн в Нью-Йорке; Профессор педиатрии Медицинской школы Дональда и Барбары Цукер в Хофстра / Нортвелл
Филлис В. Спайзер, доктор медицины, является членом следующих медицинских обществ: Американской ассоциации клинических эндокринологов, Общества эндокринологов, Общества педиатрических эндокринологов, Общества педиатрических исследований
Раскрытие информации: нечего раскрывать.
Генетическая сложность сложного заболевания
Центральный гипогонадотропный гипогонадизм (ЦГГ) — это возникающее патологическое состояние, часто связанное с избыточным весом, метаболическим синдромом, диабетом и дефектами средней линии. Генетические механизмы включают мутации по крайней мере в двадцати четырех генах, регулирующих миграцию, секрецию и активность нейронов GnRH. До сих пор механизмы, лежащие в основе CHH, как в препубертатном, так и во взрослом возрасте, остаются неизвестными в большинстве случаев.Действительно, все обнаруженные варианты генов могут объяснить небольшую долю пораженных пациентов (43%), что указывает на то, что другие гены или эпигенетические механизмы участвуют в возникновении CHH. Цель этого обзора — обобщить текущие знания о генетическом фоне CHH, упорядочить большое количество данных, представленных в литературе, в ясной и сжатой форме, чтобы создать полезное руководство, доступное для исследователей и клиницистов.
1. Введение
Физиологическая функция оси гипоталамус-гипофиз-гонад (HPG) человека основана на пульсирующем высвобождении гипоталамического гонадотропин-рилизинг-гормона (GnRH) [1].У позвоночных декапептид GnRH регулирует секрецию лютеинизирующего гормона (ЛГ) и фолликулостимулирующего гормона (ФСГ) гонадотропами передней доли гипофиза, регулируя наступление полового созревания, гаметогенез, а у женщин — эстральный / менструальный цикл [2].
В послеродовой период нейроны, секретирующие гонадолиберин, являются неотъемлемыми членами оси HPG. Во время эмбрионального развития эти клетки происходят из внецеребральной области, а именно из носовой плакоды, и мигрируют в гипоталамус, закрытый обонятельно-вомероназальными нервами (VNNs) [2, 3].Правильное развитие и скоординированная функция нейронов, секретирующих гонадолиберин, и гонадотропов необходимы для правильной активации гонад во время внутриутробной жизни и в период новорожденности (так называемое «минипубертатное созревание»). После фазы покоя в младенчестве и детстве активность HPG возобновляется во время полового созревания и на протяжении всего репродуктивного возраста взрослого человека [1].
У мужчин после рождения ось HPG активна примерно до 6 месяцев жизни, при этом концентрация гонадотропина и половых стероидов достигает пика между 4 и 12 неделями жизни.По истечении этого периода пульсация гонадолиберин снижается, и ось HPG становится неподвижной на протяжении всего детства. Начало полового созревания характеризуется пульсирующим высвобождением ГнРГ из гипоталамуса, что стимулирует секрецию ЛГ гипофизом, что, в свою очередь, стимулирует выработку тестостерона клетками Лейдига яичка [4, 5]. ГнРГ также увеличивает секрецию ФСГ, что способствует созреванию семенных канальцев и сперматогоний [5]. Адренархе в возрасте 6-7 лет начинается, когда в кровообращении начинают увеличиваться концентрации андрогенов надпочечников, ДГЭА, ДГЭАС и андростендиона, продуцируемые ретикулярной оболочкой надпочечников [5, 6].Адренархе продолжается примерно до 14 лет у обоих полов [4, 7]. Пубархе часто начинается одновременно с развитием яичек, тогда как рост волос в подмышечных впадинах происходит примерно во время максимальной скорости роста (около 14 лет) [8]. Задержку полового созревания обычно определяют как отсутствие признаков полового созревания к 14 годам у мальчиков [7].
У женщин в детстве цикл яичников, контролируемый осью HPG, находится в состоянии покоя [9]. Адренархе (возраст 6–8 лет), клинически проявляющийся ростом лобковых и подмышечных волос [7, 9], сопровождается гонадархе, и в этот период наблюдается ночной пульс ЛГ, что приводит к выработке яичниками тестостерона и прогестерона.Гонадные стероиды способствуют пубертатному скачку роста и развитию груди (телархе), который обычно начинается в возрасте 8-9 лет и приводит к менархе [7, 9]. Скачок роста происходит в 12-летнем возрасте, а затем наступает первый менструальный цикл (менархе) [7]. Менархе, вызванный ростом фолликулов и выработкой эстрадиола, возникает при начальном отделении эндометрия с последующим кровотечением. Половое созревание у женщин заканчивается, когда у женщины происходит овуляция, и повторные овуляторные циклы обеспечивают репродуктивную компетентность [9, 10].
Сложное заболевание, называемое центральным гипогонадотропным гипогонадизмом (ЦГГ), способно серьезно нарушить физиологическую функцию оси HPG у обоих полов. CHH характеризуется задержкой или отсутствием полового развития и бесплодием, связанным с несоответствующим низким уровнем гонадотропинов (ЛГ и ФСГ) и половых стероидов (тестостерон или эстрадиол) при отсутствии анатомических или функциональных аномалий оси HPG [11–13]. У пациентов с нормальным уровнем циркулирующих гонадотропинов ЛГ и ФСГ секретируются непульсирующим образом и неэффективны на целевом уровне [1].CHH — редкое заболевание, встречающееся у 1: 8000 женщин и 1: 4000 мужчин [1]. Заболевание, связанное с нормальным обонянием, встречающееся у 40–45% пациентов с ХГГ [13], называется нормосмическим идиопатическим центральным гипогонадизмом (НЦГ) или нормосмическим гипогонадотропным гипогонадизмом (НГГ). Когда нарушение обоняния, гипосмия или аносмия связаны с гипогонадотропным гипогонадизмом, у 60% пациентов [13] заболевание называется синдромом Каллмана (СК), что объясняется общим эмбриональным происхождением и путями развития ГнРГ и обонятельных нейронов. [1, 11, 12, 14].CHH бывает врожденным или приобретенным, и он может быть изолированным или сочетаться с другими гормональными дефектами гипофиза [1]. Пациенты мужского пола, пораженные CHH, часто имеют дефектную андрогенизацию и рост в периубертатном возрасте, но микропенис и крипторхизм могут проявляться уже в неонатальном периоде, что указывает на дефектную активацию HPG во время пренатального развития. У женщин обычно наблюдается первичная аменорея и задержка роста. Могут присутствовать дефекты средней линии и / или почек, которые могут быть связаны с определенными типами наследования [1, 15].
В последние два десятилетия гипогонадотропный гипогонадизм считался необратимым заболеванием, для лечения которого требовалось долгосрочное гормональное воздействие. Напротив, хорошо известно, что небольшая часть пациентов мужского пола, до 10%, после воздействия андрогенной терапии, может претерпеть обращение гипогонадизма [16]. Сообщалось, что пациенты с KS или нормосмическим HH с мутациями в рецепторе 1 фактора роста фибробластов ( FGFR1 ), KAL1 , GNRHR и CHD7 или с еще неизвестными генетическими дефектами, имели обратимый фенотип после терапии. вывод [17–21].У этих пациентов произошло спонтанное восстановление пульсирующей секреции ЛГ и нормализация уровня тестостерона после прекращения терапии [20]. Хотя точный механизм обращения гипогонадотропного гипогонадизма неясен, может быть задействована пластичность нейронов, продуцирующих GnRH, во взрослом возрасте [20]. Способность нервной системы адаптироваться к окружающей среде — поразительная особенность мозга позвоночных. Хотя нейрогенез у людей происходит в основном на эмбриональной и ранней постнатальной стадиях, мультипотенциальные клетки-предшественники в подкорковом белом веществе головного мозга взрослого человека были идентифицированы как имеющие потенциал для замены нейрональных клонов [22].Более того, нейроны обонятельного эпителия и зубчатой извилины гиппокампа генерируются на протяжении всей жизни [23-25], и их генерация, по-видимому, модулируется половыми стероидами [26]. Текущая гипотеза, объясняющая обращение гипогонадотропного гипогонадизма, предполагает действие половых стероидов на усиление пластичности нейронной сети, продуцирующей гонадолиберин, в мозге взрослого человека [20].
2. Диагностика и лечение CHH
Диагноз можно заподозрить до полового созревания, если у мальчиков имеется микропенис и / или односторонний или двусторонний крипторхизм, а также при наличии других связанных врожденных аномалий, таких как дефекты средней линии (расщелина неба, короткие пястные кости, потеря слуха, бимануальная синкинезия и др.) [1, 27]. Напротив, у новорожденных девочек нет явных отклонений от нормы, позволяющих предположить врожденный диагноз ЗГП. Возникшая у взрослых ЗГП характеризуется вторичной аменореей, снижением либидо, бесплодием и остеопорозом у женщин и симптомами снижения либидо, отсутствием утренней эрекции, эректильной дисфункцией, неспособностью к активной физической активности, депрессией, усталостью и бесплодием у мужчин [28 ].
Измерение общего утреннего тестостерона настоятельно рекомендуется в качестве первоначального диагностического теста [29], особенно для определения уровней свободного или биодоступного тестостерона [30].Несмотря на широкое использование в прошлом, практическая ценность теста GnRH была поставлена под сомнение, поскольку этот тест не предоставляет дополнительной диагностической информации по сравнению с исходными уровнями гонадотропина [28]. Функцию гипофиза можно сначала оценить по базальному гормональному уровню (измеренному с помощью сверхчувствительных анализов). Функцию щитовидной железы следует оценивать с помощью ТТГ в сочетании со свободным Т4. IGF-I можно использовать для оценки соматотропной оси, тогда как вторичный дефицит надпочечников можно оценить путем измерения утреннего кортизола и АКТГ.Аносмия может быть диагностирована путем опроса пациента и с помощью ольфактометрии в качестве теста идентификации запаха Университета Пенсильвании, полезного для определения нормального или частично дефектного обоняния [28].
Магнитно-резонансная томография (МРТ) области гипоталамус-гипофиз очень полезна при лечении ХГГ, поскольку МРТ может выявить уродства или опухоли. Ультразвуковое исследование почек обычно рекомендуется пациентам с синдромальной хронической сердечной недостаточностью, такой как синдром Каллмана. Генетическое исследование обычно является заключительным этапом исследования CHH, и полная клиническая характеристика может быть очень полезной при выборе гена / ов для скрининга [28].
Целями терапии гипогонадных подростков или молодых людей являются индукция и поддержание нормального полового созревания и индукция фертильности.
Терапия тестостероном рекомендуется взрослым мужчинам с симптоматической недостаточностью андрогенов для улучшения сексуальной функции и увеличения мышечной массы и силы. Тестостерон является основным полезным лечением для индукции и поддержания вторичных половых признаков и сексуальной функции у пораженных мужчин, но он не восстанавливает фертильность [28].В настоящее время доступно несколько составов тестостерона, таких как внутримышечные инъекции сложных эфиров тестостерона длительного действия, гелевые составы или пластыри тестостерона, применяемые каждую ночь [29]. Когда желательна фертильность, необходима терапия гонадотропинами, чтобы вызвать сперматогенез у пораженных мужчин. Обычная терапия гонадотропинами сочетает в себе хорионический гонадотропин человека (ХГЧ) и фолликулостимулирующий гормон (ФСГ) [31, 32].
3. Генетическая основа CHH
Небольшой процент пациентов (приблизительно 4%) показывает хромосомную перестройку как причину CHH или KS [7, 33], но большинство пациентов с гипогонадизмом имеют мутацию в одной или нескольких гены.
Изолированный дефицит ГнРГ, вызванный дефектами секреции или действия гипоталамического ГнРГ, является одним из редких генетических заболеваний, первоначально считавшихся строго моногенными, но многочисленные исследования этого заболевания привели к открытию нескольких новых локусов [34] ( Таблица 1), с ключевыми ролями в онтогенетическом и нейроэндокринном контроле репродукции человека [1, 12, 14]. На данный момент идентифицировано двадцать четыре гена (таблица 1).
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Идентификатор гена: идентификационный номер, присвоенный конкретному гену в базе данных NCBI; OMIM: онлайн-каталог человеческих генов и генетических нарушений, числа относятся к первому результату поиска, включая название гена и термин «гипогонадизм»; KS: синдром Каллмана; nHH: нормосмический гипогонадотропный гипогонадизм; XR: Х-сцепленный рецессивный; AR: аутосомно-рецессивный; AD: аутосомно-доминантный; OFC: синдромальная орофациальная щель; ВГА: врожденная атрезия слуха. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Белки, кодируемые генами, вовлеченными в данную патологию, были сгруппированы в зависимости от их функции по трем функциональным категориям: развитие и миграция нейронов GnRH, регуляция секреции GnRH и действие GnRH и гонадотропинов. [1, 11, 12, 35] (Рисунок 1). Однако функция некоторых генов, выявленных недавно, до конца не выяснена.
Хотя в каждом гене были идентифицированы множественные мутации, у большинства пораженных пациентов (примерно 60%) мутации не обнаруживаются, что означает, что еще предстоит обнаружить еще больше локусов болезни [14].
3.1.
KAL1
Ген KAL1 , расположенный на коротком плече хромосомы X (Xp22.3), содержит 14 экзонов и кодирует гликопротеин внеклеточного матрикса аносмин-1, который, по-видимому, участвует в миграции GnRH и обонятельной ткани. нейроны во время эмбриологического развития [36] (Рисунок 1). Гипогонадизм при СК, вызванный дефицитом ГнРГ, вероятно, является результатом нарушения миграции нейронов эмбриона, а дефектное обоняние связано с гипоплазией или аплазией обонятельных луковиц и трактов [37].Аносмин 1 представляет собой секретируемый белок, содержащий богатую цистеином область, высококонсервативный четырехдисульфидный ядерный домен белка сывороточной кислоты (WAP), четыре повтора фибронектина типа III (FNIII) (включая FNIII-1, FNIII-2, FNIII-3 и FNIII-4 repeat) и богатую гистидином С-концевую область [36].
Мутации в гене KAL1 были впервые описаны у мужчин с Х-сцепленным рецессивным KS [38]. К настоящему времени в литературе описано более 60 различных мутаций, включая делеции, миссенс, сдвиг рамки считывания [39] и грубые реаранжировки [37, 40].Около половины мутаций приходится на домен WAP и повторы FNIII, что указывает на их важную роль в правильном функционировании аносмина-1 [41]. Все мутации KAL1 составляют от 33 до 70% семейных случаев KS и от 3,1 до 27,8% явно спорадических форм известного KS [36, 37].
3.2.
FGFR1 (KAL2) и FGF8 (KAL6)
FGFR1 , кодирующий рецептор-1 фактора роста фибробластов, является членом суперсемейства рецепторных тирозинкиназ.Передача сигналов FGF контролирует пролиферацию, миграцию, дифференцировку, выживание клеток и играет важную роль в различных процессах эмбрионального развития. В присутствии протеогликанов сульфата гепарина (HSPG), FGF8 (фактор роста фибробластов-8) связывается с высоким сродством с FGFR1 и индуцирует димеризацию рецептора и его активацию [42, 43]. FGFR1 участвует в гаструляции, спецификации органов, формировании паттерна многих тканей, включая мозг, а также в развитии обонятельной системы [43] (Figure 1).
И FGFR1 , и FGF8 , отображаемые на 8p12 и 10q24, соответственно, могут вызывать аутосомно-доминантную форму KS. Большинство мутаций в FGFR1 или FGF8 являются миссенс-мутациями. Примечательно, что около 30% найденных вариантов FGFR1 являются мутациями de novo [39]. У пациентов с СК скелетные аномалии, такие как синдактилия, полидактилия или камптодактилия, наблюдались исключительно при наличии мутаций FGFR1 / FGF8 [44].
3.3.
PROK2 (KAL4) и PROKR2 (KAL3)
Ген PROK2 на хромосоме 3p13 кодирует белок прокинетецин 2, пептид из 81 аминокислоты, который передает сигнал через связанный с G-белком продукт PROKR2 ген на хромосоме 20p12.3 [45]. Аминоконцевой домен прокинетицина 2 содержит последовательность из шести аминокислотных остатков (AVITGA), которая является консервативной для всех ортологов млекопитающих и не млекопитающих. Прокинетицины могут связываться с двумя разными рецепторами, связанными с G-белком, PROKR1 и PROKR2 , с идентичностью последовательностей примерно на 85%.У них есть центральное ядро, образованное семью трансмембранными доменами (TM1-TM7), соединенными внутриклеточными и внеклеточными петлями. PROKR1 в основном экспрессируется в периферических тканях, включая эндокринные железы, тогда как PROKR2 показывает относительно локализованное распределение в центральной нервной системе, в частности в субвентрикулярной зоне (SVZ) и в обонятельных луковицах [46]. Было показано, что эта система лиганд-рецептор необходима для нормального развития обонятельной луковицы и репродуктивной системы у мышей и пациентов с ХГГ.Гомозиготные мутантные мыши с нокаутом Prokr2 демонстрируют аномальное развитие обонятельной луковицы и тяжелую атрофию репродуктивной системы, но у гетерозиготных мышей не наблюдалось значительных аномалий [37]. Модели нокаута для лиганда ( Prok2 ) или рецептора ( Prokr2 ) выявили роль в морфогенезе обонятельной луковицы и половом созревании, указывая на то, что PROK2 и PROKR2 являются сильными генами-кандидатами на дефицит человеческого GnRH [47].Мутации, описанные в системе PROK2 / PROKR2 , составляют менее 10% субъектов с KS и нормосмическим HH [37]. Нерепродуктивные, не обонятельные клинические аномалии, связанные с синдромом Каллмана, по-видимому, ограничиваются пациентами с моноаллельными мутациями [45].
3.4.
CHD7 (KAL5)
Хромодоменовый ДНК-связывающий белок 7 ( CHD7 ) на хромосоме 8 кодирует фактор ремоделирования хроматина, принадлежащий к семейству из девяти белков CHD и обладающий общей способностью использовать гидролиз АТФ для изменить структуру нуклеосомы [45, 48].Большой белок CHD7 из 2997 аминокислот содержит два важных хромодомена на своем N-конце [48]. Считалось, что хромодомены опосредуют взаимодействия хроматина, и было обнаружено, что они взаимодействуют с ДНК, РНК и гистоновыми мишенями [48].
Гетерозиготные мутации CHD7 обнаруживаются более чем у 60% пациентов с типичным синдромом CHARGE, мультисистемным аутосомно-доминантным или спорадическим заболеванием, включая колобому, сердечные аномалии, атрезию хоан, задержку развития, генитальные и ушные аномалии [49] .В литературе было обнаружено, что от 3 до 5% пациентов с хроническим гепатитом или соматическим синдромом имеют мутацию CHD7 [50].
Предыдущие исследования CHD7 показали, что анализ этого гена следует проводить у пациентов с СК, имеющими как минимум два признака синдрома ЗАРЯДА. Следуя этому руководству, Бергман и его коллеги [51] выполнили анализ CHD7 в когорте из 36 голландских пациентов с KS (ранее исключенных как носителей мутаций в FGFR1, PROK2, PROKR2 и FGF8), выявив 3 гетерозиготные мутации CHD7 и у пациентов. имеющие те же признаки синдрома ЗАРЯДА.Напротив, в исследовании Kim et al. [48], мутации в этом гене были обнаружены также у пациентов с СК, не несущих признаков ЗАРЯДА. Таким образом, с учетом этих результатов необходимы новые исследования пациентов с CHD7, и гипогонадизмом.
3.5.
NELF
NMDA рецептор синаптонуклеарная передача сигналов и фактор миграции нейронов, также известный как назальный эмбриональный фактор LHRH ( NELF ), карты на хромосоме 9. Тип наследования CHH, вероятно, будет аутосомно-рецессивным из-за двуаллельных мутаций, снижающих белок экспрессия in vitro, была описана в литературе только у одного пациента с KS без мутаций в других генах, тогда как гетерозиготные мутации NELF были обнаружены только у больных нормосмическим HH / KS пациентов с гетерозиготными мутациями в другом гене [52].
NELF — хороший ген-кандидат на роль в миграции нейронов GnRH, половом созревании млекопитающих и патофизиологии KS. Более десяти лет назад NELF мыши был клонирован из дифференциального скрининга библиотеки кДНК мигрирующих и немигрирующих нейронов GnRH [53]. Было обнаружено, что его экспрессия выравнивается вдоль плазматической мембраны обонятельных нейронов и нейронов GnRH до того, как они попадают в гипоталамус, и подавляется, когда нейроны GnRH достигают переднего мозга. Более того, снижение экспрессии белка NELF приводит к уменьшению количества нейронов GnRH и уменьшению сложности и длины нервных волокон GnRH [53].Два исследования выявили NELF в KS [54, 55]. Впоследствии эти данные были подтверждены Xu et al. [56], которые коррелировали человеческие мутации NELF с моногенным нормосмическим HH / KS. Xu и его коллеги [56] продемонстрировали, что белок NELF больше экспрессируется в мигрирующих, чем в постмиграционных нейрональных клетках GnRH, а нокдаун NELF резко ухудшает миграцию нейрональных клеток GnRH in vitro. Сигнал функциональной ядерной локализации и два предполагаемых цинковых пальца идентифицируют NELF как ядерный белок, возможно, как фактор транскрипции [56, 57].
3.6.
GNRh2 и GNRHR
GnRH играет ключевую роль в контроле репродуктивной функции (рис. 1). Он синтезируется в небольшом количестве нейронов гипоталамуса и высвобождается пульсирующим образом в портальную циркуляцию гипофиза, достигая передней доли гипофиза, где, связываясь со специфическими рецепторами (GnRHR), регулирует гонадный стероидогенез у обоих полов, стимулируя синтез и высвобождение двух гонадотропины (ЛГ и ФСГ) [58] (Рисунок 1).Человеческий GNRh2 на хромосоме 8, состоящий из четырех экзонов, разделенных тремя интронами, кодирует препропротеин из 92 аминокислот (пре-про-GnRH), состоящий из сигнальной последовательности (23 аминокислоты), за которой последовательно следуют два остатка серина, Декапептид GnRH, последовательность GKR и пептид из 56 аминокислот, названный GAP (ассоциированный с GnRH пептид) [58, 59]. Последовательность зрелого декапептида консервативна у большинства млекопитающих [60].
В двух исследованиях на мышах с делецией Gnrh2 было показано полное отсутствие синтеза GnRH [61, 62].Более того, эти мыши были неполовозрелыми, бесплодными и демонстрировали низкие уровни половых стероидов и гонадотропинов [61, 62]. Эти данные на мышах предполагают, что мутации в человеческом GNRh2 могут вызывать CHH. Варианты гена GNRh2 человека очень редки. Встречающиеся в природе мутации в человеческом GNRh3 до сих пор не описаны. Мутация p.R31C является единственной миссенс-мутацией, затрагивающей декапептидную последовательность зрелого GnRH, кодируемую GNRh2 , описанную до сих пор [60].Эта мутация, при которой аргинин замещен цистеином в положении 8 зрелого декапептида, представляет собой горячую точку [52, 59].
Другие описанные варианты были обнаружены в дополнительных декапептидных положениях, таких как p.R73X, p.T58S и p.V18M [59]. Все эти варианты были описаны в гетерозиготном состоянии, но любопытно, что Chan et al. [59] сообщили также о гомозиготной мутации p.G29GfsX12 у пациента мужского пола с тяжелым врожденным CHH. Эта делеция одной пары оснований вызывает сдвиг рамки считывания, который, как было предсказано, разрушает декапептид GnRH [59].
Ген рецептора GnRH ( GNRHR ), в отличие от того, который кодирует его лиганд, отвечает за многие инактивирующие мутации, приводящие к нарушению действия GnRH [63]. GNRHR представляет собой белок из 328 аминокислот, кодируемый геном, расположенным на хромосоме 4q21.2. Его активация приводит к увеличению активности фосфолипазы С и мобилизации внутриклеточного кальция [1]. GNRHR содержит семь трансмембранных доменов и внеклеточный амино-концевой домен из 35 аминокислот с двумя предполагаемыми сайтами гликозилирования.Интересно, что этот рецептор не имеет карбоксиконцевого цитоплазматического хвоста, поэтому он относительно медленно интернализируется и не десенсибилизируется быстро [58]. Инактивирующие мутации GNRHR были первыми, которые были признаны моногенными причинами состояния CHH [64]. Нулевые модели мышей GNRHR демонстрируют фенотип, сходный с человеческим CHH [1]. Было описано более 22 человеческих инактивирующих мутаций GNRHR без очагов [63], и эти различные генотипы приводят к широкому фенотипическому спектру, от синдрома фертильного евнуха и частичного гипогонадотропного гипогонадизма до наиболее полной формы устойчивости к GnRH, характеризующейся крипторхизмом. , микропенис, неопределяемые гонадотропины и отсутствие пубертатного развития [1].Хотя многие дефекты в большом количестве различных генов были связаны с CHH, GNRHR все еще является наиболее часто поражаемым геном при этом патогенном состоянии [64]. Поскольку подавляющее большинство пациентов с мутациями GNRHR устойчивы к GnRH, эффективное лечение бесплодия достигается с помощью гонадотропинов [63].
3,7.
LEP и LEPR
Жировая ткань посредством экспрессии и секреции лептина играет динамическую роль в энергетическом гомеостазе всего тела, действуя как эндокринный орган [65].Двадцать лет назад на мышиной модели, названной « ob / ob, », была обнаружена инактивирующая мутация гена Ob / в обоих аллелях, вызывающая полный дефицит продукта гена ob, на сегодняшний день известного как лептин ( Леп) [66]. Интересно, что избыточный вес у этих мышей с метаболическими, эндокринными и иммунными нарушениями регрессировал при введении экзогенного лептина [67].
Лептин, кодируемый человеческим геном LEP на хромосоме 7q31.3, представляет собой пептид из 167 аминокислот с мотивом пучка из четырех спиралей, аналогичным цитокину.Он продуцируется пульсирующе, следуя циркадному ритму, в основном в жировой ткани, но также и в других различных областях, таких как плацента, яичники и эпителий молочных желез [67]. Лептин и его рецептор (LEPR) присутствуют также в сперматозоидах и семенных канальцах человека [68]. В нескольких исследованиях описаны человеческие семьи с врожденным дефицитом лептина с ранним началом ожирения, гиперфагией, гипогонадотропным гипогонадизмом и задержкой полового созревания. Интересно, что инактивирующая мутация гена рецептора лептина, кодируемого человеческим LEPR на хромосоме 1p31, вызвала менее серьезные клинические проявления, указывая на то, что, вероятно, в случае дисфункции рецептора лептин способен взаимодействовать с другими молекулами, чтобы оказывать свое действие [67 , 68].На основании недавних открытий предполагается, что человеческий лептин участвует в секреции гонадолиберина посредством стимуляции нескольких нейронов гипоталамуса, секретирующих нейропептид Y, проопиомеланокортин и кисспептин 1 [67, 68]. Предполагается, что при наличии резистентности к лептину из-за ожирения лептин не может стимулировать секрецию гонадолиберина, что приводит к низким уровням ФСГ и ЛГ и гипогонадизму [67, 68].
3.8.
TAC3 и TACR3
Тахикинины млекопитающих включают семейство белков, включающее вещество P (SP), нейрокинин A (NKA), нейрокинин B (NKB) и гемокинин-1 (HK-1).Человеческие тахикинины, характеризующиеся общей С-концевой аминокислотной последовательностью (Phe-X-Gly-Leu-Met-Nh3) [69], кодируются тремя разными генами. TAC1 , TAC3, и TAC4 [70]. Ген TAC3 кодирует NKB. Исследования показывают, что эти пептиды играют роль посредников неадренергической и нехолинергической возбуждающей нейротрансмиссии, а недавние данные показывают, что тахикинины присутствуют в сперматозоидах человека и участвуют в регуляции подвижности сперматозоидов [69].Тахикинины (SP, NKA и NKB) взаимодействуют с тремя рецепторами: NK1R, NK2R и NK3R [69, 71, 72]. Специфические взаимодействия трех тахикининов с каждым из этих рецепторов и их сродство варьируются следующим образом: SP> NKA> NKB для NK1R; НКА> НКБ> СП для NK2R; NKB> NKA> SP для NK3R [71].
Конкретная система лиганд / рецептор NKB / NK3R (кодируется TAC3 / TACR3 ), ранее исследованная только для преэклампсии, алкогольной и кокаиновой зависимости [73–75], приобрела все более важную роль в репродуктивной системе человека. и хронической болезни сердца, развившейся в последние семь лет.К настоящему времени зарегистрировано более 40 пациентов с ХГГ с мутациями TAC3 и TACR3 , распространенных во всем мире [45, 76–79]. Эти пациенты характеризуются отсутствием полового созревания с низким уровнем циркулирующего сывороточного ЛГ, низким уровнем гонадных стероидов и высокой распространенностью микрофаллоса, что указывает на то, что передача сигналов NKB / NK3R важна для нормальной активации репродуктивной оси на поздних сроках беременности [45, 76– 79]. Недавние данные подтверждают, что NKB способен модулировать высвобождение гонадотропина посредством своего действия на нейроны Kiss1 [71, 80], но многие аспекты физиологии системы NKB / NK3R в контексте репродукции еще предстоит полностью охарактеризовать.
Ген TAC3 картируется на хромосоме 12q13-q21 и состоит из 7 экзонов, 5 из которых транслируются с образованием пептида препротахикинина B. Этот препропептид подвергается ферментативному расщеплению с образованием сначала пронейрокинина B, а затем зрелого NKB. Аминокислотная последовательность конечного активного пептида кодируется только экзоном 5 [81]. К настоящему времени три причинные мутации, вариант сплайсинга (c.209-1G> C) [12, 79], вариант со сдвигом рамки считывания (G20fsX39) [76] и миссенс вариант (M90T) [78], были обнаружены в Пациенты с ХГГ (таблица 2).Все эти мутации (таблица 2) произошли в гомозиготном состоянии, что свидетельствует об аутосомно-рецессивном наследовании.
| |||||||||||||||||||||||||||||||||||
Ген, кодирующий рецептор NKB ( TACR3 ) на хромосоме 4q25, состоит из пяти экзонов. Выявленные мутации, синонимичные или несинонимичные или влияющие на физиологический сплайсинг, широко распространены вдоль гена, охватывая все основные домены [12, 15, 76–78, 82] (Table 2).
3.9.
KISS1 и KISS1R (GPR54)
Кисспептин-1, кодируемый геном KISS1 на хромосоме 1q32, был рано идентифицирован в 1996 году как супрессор метастазирования при злокачественной меланоме человека [83].Ген KISS1 кодирует пептид из 54 аминокислот, также называемый кисспептином 54, который соответствует остаткам 68–121 препропротеина.
Его рецептор, KISS1R, также известный как GPR54, кодируется генным картированием на хромосоме 19p13.3. В 2009 г. инактивирующие мутации в гомозиготном состоянии в KISS1R были обнаружены у членов кровно-родственных семей с историей нормосмического HH, что выявило репродуктивную роль KISS1R и его лиганда [11]. Пока что мутации в генах, кодирующих кисспептин 1 и TAC3, а также мутации в их рецепторах (KISS1R и TACR3, соответственно.), связаны с дефицитом ГнРГ и неспособностью инициировать и / или прогрессировать в период полового созревания [84]. Инактивирующие мутации в KISS1 и KISS1R демонстрируют аутосомно-рецессивный характер передачи. На сегодняшний день зарегистрировано лишь несколько пациентов с мутациями KISS1R [85]. Благодаря экспериментам на мышах исследователи выдвинули гипотезу, что система лиганд / рецептор TAC3 / TACR3 способна регулировать высвобождение кисспептина-1, который посредством взаимодействия с рецептором KiSS-1R стимулирует высвобождение гонадолиберина и нормальную репродуктивную функцию [80]. , 84].
3.10.
PCSK1
Дефицит пропротеина / нейроэндокринной конвертазы, вызванный редкими мутациями в гене PCSK1 , был связан с ожирением, тяжелой мальабсорбционной диареей и некоторыми эндокринными аномалиями [86]. Нейроэндокринные конвертазы — это ферменты, обрабатывающие большие пептиды-предшественники для высвобождения биоактивных фрагментов, как в случае проопиомеланокортина, который процессируется нейроэндокринной конвертазой 1 (NEC 1), кодируемой геном PCSK1 на хромосоме 5q15-q21, в кортикотрофе до продуцируют адренокортикотропный гормон и липотропин [11].Первая мутация PCSK1 , сложная гетерозиготная мутация, была идентифицирована в 1997 году у пациента с ожирением и гипогонадотропным гипогонадизмом (Gly483Arg и мутация донорского сайта сплайсинга в интроне 5, вызывающая пропуск экзона 5 и преждевременное создание стоп-кодона. ) [87]. У двух других пациентов со сходным фенотипом сложная гетерозиготная мутация (Glu250X и Ala213del) и гомозиготная замена Ser307Leu были идентифицированы в PCSK1 [88, 89]. До сих пор считается, что PCSK1 действует на процессинг прогормона GnRH, даже если молекулярные механизмы все еще неясны [11, 86].
3.11.
WDR11
Ген WDR11 на хромосоме 10q26 кодирует белок из 1224 аминокислот, первоначально идентифицированный как потенциальный супрессор опухоли в клетках глиобластомы человека [90]. Идентификация человеческих мутаций WDR11 в нормосмическом HH / KS, отсутствующих в контроле, указывает на то, что WDR11 играет важную роль в половом созревании человека. К настоящему времени идентифицировано пять миссенс-мутаций (R395W, H690Q, F1150L, A435T и R448Q) [35, 91]. Четыре из них, R395W, H690Q, F1150L и A435T, полностью законсервированы во всех 11 доступных ортологах млекопитающих, что позволяет предположить, что эти замены у шести независимых, спорадических пациентов могут быть очень вредными.Все пять описанных вариантов находились в гетерозиготном состоянии, что свидетельствует об аутосомно-доминантном наследовании. Отсутствие усекающих бессмысленных мутаций и мутаций сдвига рамки считывания может указывать на более тяжелый фенотип или эмбриональную летальность [35, 91].
3.12.
HS6ST1
Hs6st1 (гепарансульфат 6-O-сульфотрансфераза 1), принадлежащий к классу молекул, участвующих в развитии нейронов, высоко экспрессируется в головном мозге [92]. Недавно было обнаружено, что ген HS6ST1 , отображаемый на хромосоме 2q21, мутирован у семи пациентов с гипогонадизмом с нормальным обонянием (nHH) или различной степенью обонятельной дисфункции (KS) [92].Все идентифицированные мутации затрагивают аминокислотные остатки, которые являются высококонсервативными в HS6ST1, но сегрегируют как сложный признак в семьях, не следуя критериям Мендели (Таблица 1). В исследовании Торнберга и соавт. [92] предполагает, что идентифицированных миссенс-мутаций HS6ST1 не могло быть достаточно, чтобы вызвать заболевание, что указывает на возможное совместное появление других мутировавших генов. Принимая во внимание важность гепарансульфата в ведении аксонов во время развития мозга у мышей [93], предполагается, что HS6ST1 играет важную роль во время развития и миграции нейронов GnRH у людей.
3.13.
SEMA3A и SEMA7A
Хорошо известно, что нейроны GnRH генерируются вне мозга, в носовой плакоде, и мигрируют по обонятельным / сошниково-носовым нервам, достигая гипоталамуса к моменту рождения. Эта миграция происходит благодаря определенным ключевым игрокам, таким как семафорины [94]. Их важная роль была усилена с идентификацией мутаций семафорина у пациентов с нейроэндокринной недостаточностью в процессе развития, связанной с бесплодием [2, 94–96].В некоторых исследованиях сообщалось, что конкретный семафорин, семафорин 3A, кодируемый человеческим SEMA3A на хромосоме 7p12.1, в случае мутации как у людей, так и у мышей, может привести к аномальной миграции нейронов GnRH в гипоталамус, что приведет к гипогонадизму и бесплодию [ 94–96] (рис. 1). К настоящему времени двенадцать различных мутаций SEMA3A , приводящих к фенотипу KS, были обнаружены у пациентов обоего пола [95–97]. Все эти варианты были идентифицированы в гетерозиготном состоянии, что указывает на аутосомно-доминантную передачу, аналогичную типу наследования, описанному у пациентов с СК с мутациями FGFR1 [39].
В 2011 году новый ген-кандидат, SEMA7A , был добавлен к уже длинному списку генов, связанных с гипогонадотропным гипогонадизмом (Таблица 1) [98]. В этом исследовании SEMA7A участвовал в нормальном развитии системы GnRh2 у мышей и был предложен как сильный генетический маркер некоторых форм дефицита GnRh2 у людей. Впервые в недавнем исследовании мутации гена SEMA7A , картированного на хромосоме человека 15q22.3-q23, были обнаружены у двух пациентов с гипогонадией, у мужчины с nHH, несущего также мутацию KISS1 и самец с KS, несущий также мутацию KAL1 [97].На основании этих результатов Кянсакоски и его коллеги предполагают, что выявленных мутаций недостаточно, чтобы вызвать патологию. Следовательно, предполагается ди / олигогенное наследование [97].
3.14.
LHB и FSHB
Гонадотропины представляют собой этеродимеры, состоящие из α-субъединицы (общей для ТТГ, ФСГ, ЛГ и ХГЧ) и конкретной β -субъединицы. До сих пор не было выявлено мутаций в гене CGA , кодирующем α-субъединицу, тогда как у некоторых пациентов мужского и женского пола с задержкой полового созревания было обнаружено несколько мутаций в генах, кодирующих β -субъединицы ЛГ и ФСГ [99, 100].
Гомозиготные мутации в гене LHB (хромосома 19q13.32), отменяющие активность ЛГ, на сегодняшний день зарегистрированы у семи мужчин и двух женщин [101–104]. У больных мужчин половая дифференцировка нормальна, но отсутствие ЛГ изменяет пролиферацию и созревание клеток Лейдига, нарушая сперматогенез [101]. Женщины с инактивирующей мутацией LHB имеют нормальное пубертатное развитие и менархе, за которыми следуют олигоменорея и вторичная аменорея [101].Немногочисленные данные, представленные в литературе, описывающие мутации LHB , предполагают, что одной копии LH бета достаточно для нормальной секреции LH и функции гонадотропной оси, действительно, только пациенты, несущие гомозиготные мутации, демонстрировали гипогонадизм, в то время как их родственники, несущие гетерозиготный вариант, клинических проявлений не обнаружил [101, 102].
Ген FSHB , расположенный на хромосоме 11p13, состоит из трех экзонов, но только экзоны 2 и 3 кодируют зрелый пептид.К настоящему времени четыре различных мутации FSHB были описаны у четырех неродственных пациентов женского пола с гипогонадизмом, а три мутации описаны у трех пациентов мужского пола с CHH [99, 100]. У пораженных женщин наблюдались задержка полового созревания, отсутствие или плохое развитие груди и первичная аменорея. После лечения экзогенным ФСГ созревание фолликулов, овуляция и фертильность были достигнуты у двух женщин. У всех пораженных мужчин были маленькие яички и азооспермия, но только у одного мужчины не было пубертатного развития [99, 100].Литературные данные позволяют предположить, что наличие неопределяемого сывороточного ФСГ и высоких уровней ЛГ у пациентов с ХГГ обоего пола может быть в значительной степени обусловлено молекулярными дефектами в гене FSHB [99, 100].
3.15.
NDN
Некдин принадлежит к суперсемейству белков MAGE и способен активировать экспрессию GnRH и развитие нейронов GnRH у грызунов [105]. Некдин человека, кодируемый геном NDN (хромосома 15), потенциально играет роль в возникновении гипогонадизма у пациентов с синдромом Прадера-Вилли [106].Несколько лет назад Beneduzzi и его коллеги [105] идентифицировали редкий вариант некдина, связанный с мутацией в FGFR1 , у пациента с семейным KS. Тем не менее, функциональные исследования показали, что мутировавший некдин способен активировать экспрессию GnRH как белка дикого типа [105]. Необходимы дальнейшие исследования, чтобы выяснить роль этого белка в половом созревании и репродукции человека.
3.16.
TSHZ1
Как упоминалось ранее, субъекты CHH могут демонстрировать аномалии развития, такие как волчья пасть, потеря слуха и другие дефекты средней линии.Однако неясно, какой ген, ассоциированный с CHH, участвует и в какой степени в определении этих дефектов развития.
В 2007 году инактивация мышиного гена Tshz1 продемонстрировала его роль в развитии мягкого неба, осевого скелета и среднего уха у мышей, предполагая участие человеческого гена TSHZ1 (Teashirt Zinc Finger Homeobox 1) у пациентов с аномалиями неба, скелета и ушей [107]. Несколько лет спустя при исследовании небольшого числа лиц, страдающих синдромальной орофациальной щелью (OFC), аномалиями неба и врожденной атрезией слуха (CAA), были обнаружены мутации в гене TSHZ1 , показывающие аутосомно-доминантную сегрегацию [108, 109] .
Совсем недавно Ragancokova и его коллеги исследовали роль Tshz1 у мышей, показав, что инактивация этого гена может вызывать гипоплазию обонятельной луковицы и серьезный обонятельный дефицит [110]. Кроме того, в этом исследовании оценивали обоняние у пациентов с гетерозиготными мутациями TSHZ1 , пораженных САА, и у них была выявлена гипосмия [110]. Дальнейший анализ экспрессии генов показал ключевую роль TSHZ1 в регуляции экспрессии PROKR2 , которая связана с синдромом Каллмана [110].
Принимая во внимание эти данные, предполагается, что ген TSHZ1 участвует, прямо или косвенно, и может быть включен в группу генов, ассоциированных с CHH (Таблица 1).
4. Дигенное и олигогенное наследование
Долгое время ХГГ считались моногенной патологией с менделевской наследственностью. В 2006 году исследователи начали думать о гипогонадотропном гипогонадизме как о пищеварительном расстройстве. С этого времени другие исследователи описали случаи дигенных мутаций в нормосмическом HH или KS [14, 52].В самом деле, дефекты в разных генах могут действовать синергетически, индуцируя фенотип CHH / KS или изменяя тяжесть дефицита GnRH [15, 52]. В 2010 году Sykiotis et al. [14] идентифицировали 10 пациентов с CHH, несущих редкие варианты, изменяющие дигенный белок, и 18 пациентов с CHH, несущих известные или предсказанные олигогенные вредные мутации. В 2011 году Quaynor et al. [52] внесли свой вклад в эти данные, описывающие 48 пациентов с нормосмическим ХГГ, проверенных на 13 связанных с заболеванием генов. У 12,5% этих больных были выявлены дигенные мутации.Более того, Quaynor и его коллеги [52] предположили, что часть изолированного дефицита GnRH может быть отнесена, помимо дигенного / олигогенного компонента, к негенетическим компонентам, как в случаях случайного начала заболевания у взрослых после нормального полового созревания и репродуктивной функции у взрослых. предметы без мутаций.
Тем не менее, литературные данные указывают на то, что моногенные мутации являются причиной большинства случаев ХГГ (более 80%) [14, 15, 52].
5. Выводы
В заключение, сообщалось о генетических изменениях в двадцати четырех различных генах, описанных до сих пор в литературе и связанных с центральным гипогонадотропным гипогонадизмом.Все эти причинные варианты составляют только 40–45% пораженных пациентов, что предполагает участие других локусов и / или эпигенетических механизмов. Из опубликованных данных еще более очевидна олигогенная природа заболевания.
Учитывая сложность CHH, мы считаем, что наилучшим подходом к генетическим исследованиям могло бы стать использование лабораторных методов, таких как секвенирование следующего поколения (NGS), позволяющее проводить одновременный скрининг многих генов.
До настоящего времени мало внимания уделялось множеству непатогенных однонуклеотидных полиморфизмов (SNP), обнаруженных в этих генах.Хотя полиморфные варианты, описанные в литературе, не считаются патологическими, они могут влиять на фенотип, особенно если встречаются в сочетании с другими мутациями в других генах. Поэтому мы считаем, что их роль не следует недооценивать и необходимо провести будущие исследования корреляции полиморфизмов, причинных мутаций (если таковые имеются) и клинических характеристик пациентов.
Конфликт интересов
Авторы заявляют об отсутствии конфликта интересов в отношении публикации данной статьи.
Благодарности
Авторы участвуют в Итальянской исследовательской группе ICH, входящей в состав итальянских обществ эндокринологии и детской эндокринологии и диабета. Эта работа поддержана грантом Министерства здравоохранения Италии (грант № GR-2008-1137632) и грантом PRIN (проект «AMLET», грант № 2010C8ERKX).
Успешная беременность и живорождение от женщины с гипогонадотропным гипогонадизмом с низкими концентрациями эстрадиола в сыворотке крови, несмотря на многочисленные созревания ооцитов: клинический случай | BMC Беременность и роды
История болезни пациентки
Пациентка, 26-летняя японка, не имела беременности.Первая менструация произошла в 13 лет, и после этого менструальный цикл был регулярным. В 21 год у пациентки началась аменорея, в результате которой за два месяца похудела на 12 кг. В другой больнице, начиная с 23-летнего возраста, этому пациенту в течение двух лет была назначена пероральная заместительная терапия E 2 и P 4 . Пациентка вышла замуж в 26 лет и решила пройти курс лечения бесплодия в нашей больнице. Индекс массы тела (ИМТ) пациентки составлял 19,2 кг / м 2 , и ее женские характеристики выглядели нормальными.Диаметр правого яичника 26,0 мм, левого 29,1 мм. Концентрации ФСГ и ЛГ в сыворотке составляли 3,2 мМЕ / мл и 0,5 мМЕ / мл соответственно. Результаты ее эндокринных тестов были в пределах нормы нашей больницы, за исключением трийодтиронина (T 3 ) и пролактина (PRL) (дополнительный файл 1). Пациентке был поставлен диагноз гипоталамической аменореи после нагрузки лютеинизирующим гормон-рилизинг-гормоном (ЛГ-РГ). контрольная работа.
И яйцеводы, и просвет матки у этой пациентки были морфологически нормальными при обследовании с помощью гистеросальпингографии (HSG).Качество спермы мужа выглядело нормальным и соответствовало критериям Всемирной организации здравоохранения 2010 года [5]. При трансвагинальном ультразвуковом исследовании в обоих яичниках было обнаружено более пяти антральных фолликулов (т.е. 5–10 мм) каждый. Концентрация анти-мюллерова гормона (АМГ) в сыворотке крови составляла 10,8 нг / мл, на основании чего предполагалось, что резерв яичников функционирует [6,7,8,9]. Чтобы пройти плановое лечение бесплодия для этой пары, которая жила отдельно, сперма мужа была заморожена.
В нашей стране, однако, рекомбинантный ЛГ (рЛГ) еще не доступен, а ГМГ и хорионический гонадотропин человека (ХГЧ) должны вводиться внутримышечно только врачом или медсестрой. Из-за своего рабочего назначения и рабочей нагрузки она не могла получать ежедневные введения HMG или hCG, поэтому самостоятельное введение rFSH было только тем, что мы могли предложить этой пациентке в период COS. Количество антральных фолликулов (AFCs) и концентрация AMH у пациентки были в группе высокого риска синдрома гиперстимуляции яичников (OHSS) [10, 11].Поэтому была запланирована схема приема антагонистов гонадотропин-рилизинг-гормона (ГнРГ), чтобы избежать быстрого роста сывороточных концентраций E 2 [12, 13], только если наблюдалось надлежащее повышение сывороточных концентраций E 2 и размер доминирующих фолликулов достигал более 12–14 мм в диаметре.
Первая стимуляция яичников
Когда у этой пациентки возникло кровотечение отмены после ежедневного приема оральных контрацептивов (ОК) в течение 14 дней, измеримые фолликулы не наблюдались ни в одном яичнике на 3-й день (рис.1). В тот же день была начата COS с ежедневного введения 225 МЕ рФСГ. Наблюдалось развитие фолликулов и повышенные концентрации P 4 , но концентрации E 2 были низкими на 11-й день (Таблица 1). Режим антагонистов GnRH не применялся, поскольку не наблюдалось надлежащего повышения концентрации E 2 в сыворотке. Хотя на 13-й день в обоих яичниках было более 30 фолликулов размером 15–19 мм, сывороточные концентрации E 2 были низкими — 484 пг / мл, а концентрации P 4 — высокими на 2.09 нг / мл. COS был отменен из-за концентрации гормона, которая не отражала таковую при многочисленных фолликулярных образованиях.
Рис. 1
Развитие фолликулов после контролируемой стимуляции яичников (COS). Ответ яичников контролировали с помощью трансвагинального ультразвукового исследования во время COS. Во время первого COS не наблюдали измеряемых фолликулов ни в одном яичнике на 3-й день менструального цикла (MC). В тот же день начинали COS с ежедневного введения 225 международных единиц (МЕ) рекомбинантного фолликулостимулирующего гормона (рФСГ).Наблюдались фолликулярное развитие и повышенная концентрация прогестерона (P 4 ), но концентрации эстрадиола (E 2 ) были низкими на 11-й день. Хотя в правом (R) и левом (L) яичниках было более 30 фолликулов 15-19 мм на 13 день, сывороточные концентрации E 2 были низкими — 484 пг / мл, а концентрации P 4 были высокими — 2,09 нг / мл. COS был отменен из-за концентрации гормона, которая не отражала таковую при многочисленных фолликулярных образованиях. Со дня отмены первого COS пациент принимал 10 мг синтетического прогестерона (SP) перорально в течение следующих 14 дней.У пациентки было кровотечение отмены, и на 3-й день в обоих яичниках наблюдались несколько небольших фолликулов. С того же дня вводилась суточная доза 175 МЕ рФСГ в течение первых 5 дней, а затем 200 МЕ в течение оставшегося периода лечения. Подобно реакции, наблюдаемой при первом COS, наблюдались высокие сывороточные концентрации P 4 , 2,11 нг / мл; тем не менее, сывороточные концентрации E 2 , 701 пг / мл, не отражали концентрации многочисленных фолликулярных образований
Таблица 1 Изменение концентраций стероидных гормонов в сыворотке и развитие фолликулов во время контролируемой стимуляции яичников
Вторая контролируемая стимуляция яичников
Со дня отмены первого COS пациентка принимала 10 мг синтетического прогестерона (SP) перорально в течение следующих 14 дней.У пациентки было кровотечение отмены, и на 3-й день в обоих яичниках было обнаружено несколько небольших фолликулов (рис. 1). С того же дня вводили дневную дозу 175 МЕ рФСГ в течение первых 5 дней, а затем 200 МЕ в течение оставшегося периода лечения. Подобно реакции, наблюдаемой при первом COS, наблюдались высокие сывороточные концентрации P 4 ; однако сывороточные концентрации E 2 не увеличивались на 15 день (таблица 1).
Таким образом, во время COS мы сообщили этой пациентке, что вспомогательные репродуктивные технологии (ВРТ) могут потерпеть неудачу из-за необычных концентраций стероидных гормонов в сыворотке крови, низкого E 2 и высокого P 4 .В то же время мы также проинформировали пациента, что АРТ может быть успешной, потому что сывороточные концентрации АМГ были адекватными и в ответ на рФСГ развились многочисленные фолликулы. Пациент и муж добровольно согласились пройти АРТ, понимая процедуру с информированного согласия. В тот же день овуляцию индуцировали 5000 МЕ ХГЧ.
ЭКО / ИКСИ, культивирование эмбрионов и криоконсервация
Через 35 часов после введения ХГЧ фолликулы укололи трансвагинальной иглой для извлечения ооцитов (OPU).Аспирировали фолликулярную жидкость (ФЖ) из самого большого фолликула, которую замораживали для последующего анализа на гормоны. Криоконсервированные сперматозоиды размораживали, и они имели характеристики 44,0 × 10 6 / мл и подвижность 9,5%. Ранее сообщалось, что ооциты из низких концентраций E 2 в сыворотке пациента с ГГ снизили частоту оплодотворения при ЭКО [2]. Чтобы получить как можно больше оплодотворенных ооцитов с замороженной-размороженной спермой достаточного качества, в дополнение к ЭКО была проведена также ИКСИ.18 и 15 КОК были подвергнуты ЭКО и ИКСИ соответственно. ЭКО-КОК оплодотворяли 5,0 × 10 4 подвижных сперматозоидов / КОК. Через 16–18 ч после ЭКО и ИКСИ оплодотворение определяли по наличию двух пронуклеусов (ПН).